
Question 38: What measurements and criteria do you use to decide when to change your gas and liquid chloride absorber material? How do you determine the selection of absorber material?
John Clower (Chevron)
For both gas and liquid service, Chevron monitors the inlet HCL/Total Chloride and replaces the adsorbent/molecular sieve based on material balance loading of chloride on the adsorber media. Chevron does monitor adsorbent outlet HCL/Total Chlorides, but as a best practice will change the adsorbent material before vendor maximum loading if breakthrough has not occurred. Spent adsorbent will become acidic and pass chloride as organic chloride to the downstream processes. Organic chlorides are difficult to detect by conventional tubes in gas service and will form HCL in downstream processing units.
This performance-based approach is not without problems, e.g., the accuracy of both chloride measurements and represented adsorbent capacity, and therefore requires a trial-and-error
approach.
Represented capacity of any chloride trap material will have been set the vendor to minimize high acidity conditions that lead to organic chloride and polymer (red/green oil) production. Commercially there are four main types of chloride adsorbent material available:
•Alumina
•Modified/Promoted Alumina
•Molecular Sieve
•Metal Oxide
Each of these materials is used in Chevron Refineries and joint ventures. Each adsorbent type will have various properties that can be used in making a decision on application:
•Total chloride capacity (HCL and Organic)
•Reactivity – potential for organic chloride and red/green oil formation
•Interferences (e.g., Sulfur)
•Cost per pound of chloride removed
Also, the design of the vessel used is important (L/D for adequate flow distribution, contact time) and can result in shorten life versus predicted breakthrough. Selection of adsorbent versus service will usually be made on a cost per pound of chloride removed.

Janel Ruby (Johnson Matthey Catalysts)
Chloride can be removed from streams using various products. These chloride guard products can differ in the way they are manufactured and in the way they work in certain applications, so it is important to choose the right one for your needs. The most common products are chemical absorbents or promoted alumina adsorbents. Chemical absorbents remove chlorides by irreversible chemical reaction, meaning that the chloride is chemically bound within the absorbent. Chloride removal in promoted alumina is accomplished mainly by adsorption in which hydrogen chloride is adsorbed onto the alumina surface. Both types of beds are non-regenerable and require change-out at chloride breakthrough.
When determining which product is right for a particular service, it is important to evaluate the operating parameters of the chloride guard bed. The location of the bed in the reforming flow sheet, the operating temperature of the bed, and the normal and maximum inlet chloride levels are important factors to consider when selecting an absorbent type.
Promoted alumina products are available for liquid and gas services. Promoted alumina can work over a range of operating temperatures but chlorides that are adsorbed onto the material may desorb at higher temperatures which will decrease the effectiveness of the product in these regimes. These products also have a lower chloride capacity usually ranging from 12 to 15% wt/wt, and require a high change-out frequency. An area of concern when utilizing promoted alumina materials is the formation of undesirable side products. When the chloride binds to the alumina surface of the guard material, it creates surface acid sites. The acidic surface of the material can catalyze side reactions and lead to the creation of organic chlorides or high-molecular weight hydrocarbons called “green oils.” Green-oils not only foul equipment, but also the guard bed itself, which can cause difficulties in bed discharge (increased purge time) and disposal.
Chemical absorbents are the most favorable option for chloride removal. These products are available for use in liquid and gas services. Chemical absorbents work over a wide range of temperatures. These products have high chloride pick-ups, for example PURASPECJM 2250 is a mixed metal oxide chemical absorbent which can achieve a chloride capacity of 30% wt/wt in non-fouling, gas phase applications. As previously stated, these products remove chloride through an irreversible chemical reaction. The alumina structure present in these types of chemical absorbents acts only as a binder which minimizes the tendency for unwanted side reactions. PURASPECJM 2250 can commonly be employed with the use of just a single guard bed.
There a few other considerations surrounding chloride guard bed materials. It is important to avoid two-phase flow in these beds as this will affect the performance of the chloride guard. Both promoted alumina products and chemical absorbents have a higher pick-up in gas phase, non-fouling and non-wetting applications. In liquid applications, diffusion through the liquid film around the chloride guard particle is the rate limiting step and capacities are generally lower than gas phase duties because of the mass transfer effects. Chemical absorbent products, PURASPECJM 6250 and PURASPECJM 6255 were designed to address this concern. These products have a high capacity and specific pore structure to allow improved removal capacity. They are comprised of the same chemical formulation and micromeritic properties but represent two differing particle sizes; PURASPECJM6255 is manufactured as a smaller sized sphere. The smaller size provides better performance as this minimizes the liquid film through which the HCl must diffuse, reducing the depth of the mass transfer zone and leads to higher average chloride pick at the point of HCl breakthrough.
The presence of HCl or organo-chlorides (RCl) in the exit stream of the chloride guard bed will indicate it is time to change out the material. The life of the guard depends on how the bed(s) is configured and what type of product(s) has been installed. Unless the bed needs to be shut down for inspection or is involved in a larger turnaround plan, chloride breakthrough will be the main reason for a shutdown to replace product. Regular testing for chlorides in the exit stream will help to determine when change out is needed. In applications with longer life cycles (years) testing may only be needed monthly until the bed is getting closer to its expected change-out interval. In applications with shorter life expectancies (months), the frequency of testing should be at least weekly.
Throughout the life of the bed, it is important to measure the HCl and RCl levels both inlet and exit the chloride guard beds. It has been shown that when promoted alumina is used for HCl removal, it catalyses the conversion of HCl to organic chloride species that can then slip from the bed. If the operator is only measuring for HCl then this chloride slip can go undetected until downstream issues occur. Chlorides passing through the bed can cause corrosion of downstream equipment and formation of ammonium chloride that cause fouling and blocking of equipment e.g., stabilizer columns, exchangers and compressors.
Year
2010
Process
Question 14: What is industry experience of using tri-metal (platinum-rhenium with promoter) catalysts?
MELDRUM (Phillips 66)
Promoted or multi-metallic reformer catalysts have been a topic of research since at least the early 1970s. They have been tried commercially in various forms over the years, all with the objective of improving yields by suppressing the demetallization reactions. The current promoted catalysts have advanced the formulation of manufacturing techniques to new levels of performance. Recently, Phillips 66 has selected promoted catalysts for future reloads in at least three of our sites. The additional cost of the catalyst is justified when considering increased product yield and improved activity that allows a lower reactor temperature requirement, which both provide for a very quick payback on the additional catalyst cost.
The example shown on the slide indicates the additional yields – both in the C5+, as well as hydrogen – and some improved activity that might be expected with a promoted catalyst. When selecting the promoted catalyst, regeneration procedures should be reviewed with the catalyst vendor to ensure that maximum catalyst performance from regeneration to regeneration is achieved, particularly in the area of reduction and dryout steps.

BULLEN (UOP LLC, A Honeywell Company)
We have two catalysts that we offer in the semi-regenerator market and also for cyclic reforming applications. One of them is the R-98 catalyst that was introduced in 2005 and which has over 50 installed applications. We have a new catalyst called R500 that has better activity and stability, and we have put it in 10 units. As Craig said, proper regeneration procedures are very important for any semi-regeneration unit, and maybe even more so for these tri-metallic systems, because of the issues related to dryout and reduction. It is important to get consistency with this procedure because you will lose the advantage of the tri-metallic system if you do not do the dryout and reduction correctly. Getting that repeatability is very important.
CRAIG MELDRUM (Phillips 66)
Regeneration procedures should be reviewed with the catalyst vendor to ensure maximum catalyst performance from regeneration to regeneration. For example, UOP R-72 was a promoted catalyst offered about 15 years ago and required a different reduction procedure than the non-promoted catalyst for hydrogen concentration, pressure, temperature, and dry-down schedule.
PATRICK BULLEN (UOP LLC, A Honeywell Company)
Trimetallic catalysts containing rhenium are typical for use in fixed-bed reforming applications, both semi-regenerative and cyclic reforming applications. In recent years, both additional metals and oxides have been added to platinum-rhenium reforming catalysts. Metal promoters have been added to increase selectivity and product yields. The additional metal partially suppresses platinum-rhenium activity, reducing metal-catalyzed hydrogenolysis that lowers selectivity.
Over the past decade, UOP successfully developed the proper catalyst base, formulations (including promoter type), and manufacturing techniques needed to generate catalysts that demonstrate excellent yield stability and regenerability. UOP’s R-98 catalyst was introduced in 2005 and has over 50 successful applications with many regeneration cycles, and our customers are benefiting from the higher yields. UOP recently introduced a new product, R-500, that shows even great activity and stability, with over 10 commercial applications. It is well suited for reforming units where even longer cycle lengths are desirable or where higher activity is needed to push more barrels. The gradual acceptance of promoted catalysts is analogous to that of the bimetallic catalysts having higher rhenium content that preceded them in this market.
Proper regeneration procedures are critical for the success of any semi-regeneration catalyst; and in particular, promoted formulations that have reduced metal activity. One Best Practice is to ensure proper dry-down, reduction, and sulfiding. Cyclic reforming applications are a little more demanding due to the regeneration environment (higher moisture and sulfur, for example), but new promoted formulations have been demonstrated in these applications as well.
SONI OYEKAN (Prafis Energy Solutions)
This question needs some more definition to elicit appropriate responses with respect to what is truly a “trimetallic” catalyst. My initial response is that my experiences in the use of “trimetallic” platinum-rhenium catalysts for fixed-bed cyclic regeneration reformer operations were good. The catalysts performed as projected by the catalyst and technology supplier for catalysts containing a third metal that was specifically added for modifying the acidic functionality of the catalysts.
Having written that, it is important to understand the type of catalysts commonly referred to as “trimetallic” catalysts. The term could cover Pt/Re (platinum/rhenium) catalysts with a third metal as a modifier for the alumina to moderate the acidic functionality of the catalysts or those in which the third metals are added to modify the hydrogenation functionality of the platinum or to moderate rhenium hydrocracking activity. In other trimetallic catalyst formulations, the third metal can work in conjunction with the rhenium as co-promoters for the platinum functionality.
The performance objective of the third metal is crucial in order to assess long-term performance and benefits of the third metal. Metals on catalytic reformer catalysts typically undergo varying degrees of reduction to different oxidation states at different temperatures and adequate metals redispersion are achieved at different oxidative conditions. Trimetallic catalysts’ expected performances and potential limitations should be well understood by oil refiners before acquiring them for use. Catalyst suppliers should provide test data to show multiple regenerations and adequate reactivations of the three metals, even if the other two metals are acting as co-promoters for the platinum. Another key factor is to ensure that optimal metals distributions are achieved during catalyst manufacture. There are other factors to consider that are beyond inclusion in this short response on trimetallic catalysts.
If the third metal has been added to moderate catalyst acidic functionality and reactivation of that third metal is not an important factor other than decoking, then the refiners’ challenges are lessened to some extent. It should be recalled, however, that the history of catalytic reforming is dotted with an oil refiner’s experiences with second metals that had been added to the platinum and which led to significant performance problems. The problems were related to inadequate metals activation, especially poor redispersion of the promoter metals, and these problems led to poor catalyst performance for subsequent cycles after the first cycle for fixed-bed catalytic reforming systems. Furthermore, in reforming catalyst development programs, the addition of metals to Pt/Re catalysts led to increased feed sulfur sensitivity challenges for the resultant trimetallic catalysts. Feed sulfur sensitivity and catalyst regeneration challenges should be studied sufficiently by the catalyst and technology supplier during that supplier’s catalyst development studies leading to the production of “trimetallic” catalysts.
Year
2013
Process
Question 35: When processing tight oil crudes, are lower bed pressure drop problems in VGO/resid hydrotreater reactors a concern? If so, what mechanisms explain this issue?
LIOLIOS (DuPont Clean Technologies)
The highly paraffinic nature of the tight crudes and the destabilization of asphaltene molecules can cause precipitation and agglomeration. One of our customers with a gas oil mild hydrocracker switched feedstock to increase amounts of black wax crude. This was a five-reactor system. A guard bed reactor was first, followed by four other reactor beds. In the polishing reactor bed, this customer saw an increase in pressure drop. It was theorized that this pressure drop was caused by asphaltene precipitation and polymerization in the bed.

The following graphs show some of what was happening at this unit. It is a constant feedstock. They raised the temperature to get some additional cracking. You will notice an elevated pressure drop in the last bed shortly after they increased the severity of the unit. If you look at the next chart, you can see where they decreased the severity of operation of the unit and the pressure drop recovered. Our theory is that there was a recombination of those asphaltenes.

SHARPE (Flint Hills Resources, LP)
We have had no second and third bed ∆T problems when running high rates of Eagle Ford crude. When there were high bed ∆Ps in the lower treating beds, they were usually a result of coke fouling due to hydrogen starvation, and low hydrogen partial pressure.

GLENN LIOLIOS (DuPont Clean Technologies)
The highly paraffinic nature of tight oil crudes, and the potential increase in asphaltene precipitation when these crudes or cuts of these are mixed with polar asphaltenic oils or cuts, has been well documented. The increase in paraffin content can lead to destabilization of the asphaltene core which can then agglomerate to form larger macromolecules that may precipitate out under hydrotreating conditions.
A number of published documents2 detail the causes and reactions behind this phenomenon and outline methods to determine which crude type and cuts are compatible and what ratios are required to minimize the chance of this phenomenon occurring.
Much of the industry experience indicates that asphaltene precipitation and fouling in process units normally occurs in regions of high heat flux when agglomerated asphaltenes easily crack or dehydrogenate leaving coke-like deposits such as feed/effluent exchangers or where hydrotreater reactions are initiated; i.e., the top bed of a hydrotreating reactor. However, it was observed that a gas oil mild hydrocracking unit experienced a noticeable increase in pressure in a final polishing reactor after the feed to the unit was switched to process a feed that had been mixed with an increased percentage of highly paraffinic (black wax crude) feedstock. At the same time, the severity was increased by lowering the throughput without reducing inlet temperatures. The polishing reactor was the last in a series of five reactor beds, the bed being a separate bed reactor. During the observed increased pressure drop in the polishing reactor, no appreciable pressure drop was observed in the guard bed or main reactor beds. It is important to point out that after the space velocity and feedstock to the system were normalized, the pressure drop decreased almost to the baseline range prior to the event.
It is theorized that the observed bed pressure drop increase in the last bed was a result of asphaltene precipitation and polymerization on the bed that occurred after increased severity reactions cracked the smaller molecules that kept the increased asphaltenes in solution. According to work conducted by Wiehe on asphaltene precipitation3 , asphaltenes are maintained in solution in oil by a micelle type of configuration. This theory has been also explained by other authors4 . The asphaltene core is surrounded by a solvated shell that consists of resins. Resins are molecules with aromatic and naphthenic rings.
Under high severity conditions such as those experienced in this mild hydrocracker operation, the resins can crack into smaller molecules. This can disrupt the micelle type configurations at which asphaltenes are kept in solution, and the asphaltenes can precipitate upon cooling.
Analytical tests carried out on the hydrocarbon feed samples indicated that the asphaltene content (heptane insolubles), although low in comparison with a heavy residue5, was found to be approximately three times higher than the one on the sweet GO FCC feed sample that was being recirculated to the unit and the regular GO sample fed to the GHC.
This theory explains why the upstream reactor beds did not experience a corresponding increase in pressure drop. If it were due to deposits, catalyst fines, or simply rust from upstream units, the first two reactors should have acted as filters preventing the last bed from getting plugged-up.
JUAN ESTRADA (Criterion Catalysts & Technologies)
Two primary mechanisms for pressure drop in bottom beds are coking and asphaltene precipitation. Coking results from operation at elevated temperatures and hydrogen deficiency. Asphaltene precipitation results from a reduction in liquid solvency. The design of VGO hydrotreaters with elevated pressure, low space velocity, and high treat gas rates helps minimize coking; however, elevated saturation of aromatics reduces the solvency of the oil, increasing the potential for asphaltene precipitation in the catalyst bed.
Processing tight oils in the crude diet reduces the aromatic content of the gas oils. For this reason, the coking potential of the feed is lowered, but the potential for asphaltene precipitation increases. With lower feed aromatics and severe hydrotreatment, the solvency change may be sufficient in the lower catalyst beds to precipitate asphaltenes introduced with the other gas oil components from conventional or synthetic-derived crude sources.
The mechanism of asphaltene precipitation from a reduction in liquid solvency has been connected to many historical pressure drop problems involving changes in operation and feedstock qualities such as aromatic and C7/C9 asphaltene contents and the distillation tail. Applying this accepted mechanism to lower bed pressure drop problems in units processing tight oil derived gas oils logically explains recent pressure drop problems in a few VGO hydrotreaters. Refiners continue to learn compatibility limitations of co-processing tight oils in the crude diet, including impacts on VGO reactor pressure drop growth has become a consideration.
Year
2013
Process
Question 16: What is the typical carbon monoxide (CO) concentration in the reformer net gas? How is the CO content measured? What are the potential effects to downstream units from the CO?
MELDRUM (Phillips 66)
Carbon monoxide can form in reformer units as the hydrocarbon reacts with moisture under very low-unit pressure conditions. Typically, semi-regeneration reformer net gas would have nil CO and only a minimal amount in a CCR-type unit. I expect it to probably be on the order of 5 ppm (parts per million), though some units report routine measurements of 10 to 20 ppm CO in their net hydrogen off gas.
One of our cyclic units that was operating at 400-pound had CO as high as 20 ppm in its net hydrogen stream when the recycled moisture rose to around 300 ppm. The excessive water entered the reformer from a leaking side reboiler on a wet debutanizer that used a slipstream of the reformer reactor effluent as the heat source. The water then returned to the reformer product separator. The high CO caused deactivation in the catalyst in a downstream isomerization unit.
Accurate measurements of CO in the net gas are difficult. Reformer units are not expected to have much CO, so they seldom have an online analyzer. A colorimetric tube – Gastec or Dräger type – can be used to give an indication of the presence of CO, but accuracy for a quantified number is difficult and requires the use of a carbon pre-tube to remove the hydrocarbons.
CO is detrimental to downstream hydrogen-using units in three principal areas. CO in hydrogen being fed to a distillate hydrotreater will methanate, consuming the hydrogen that would have otherwise been used for the desulfurization reactions. This will have the effect of lower catalyst activity. CO in hydrogen fed as a makeup stream to an isomerization unit will also methanate and form moisture that will deactivate the isomerization catalyst. CO that did not methanate in the second example could act as a poison to the platinum metal function of the isomerization catalyst. UOP suggests a CO limit of 1 ppm max for isomerization hydrogen makeup gas. My Answer Book response also includes some of the common steps used to minimize CO formation in reformer units, particularly in a CCR unit.
BULLEN (UOP LLC, A Honeywell Company)
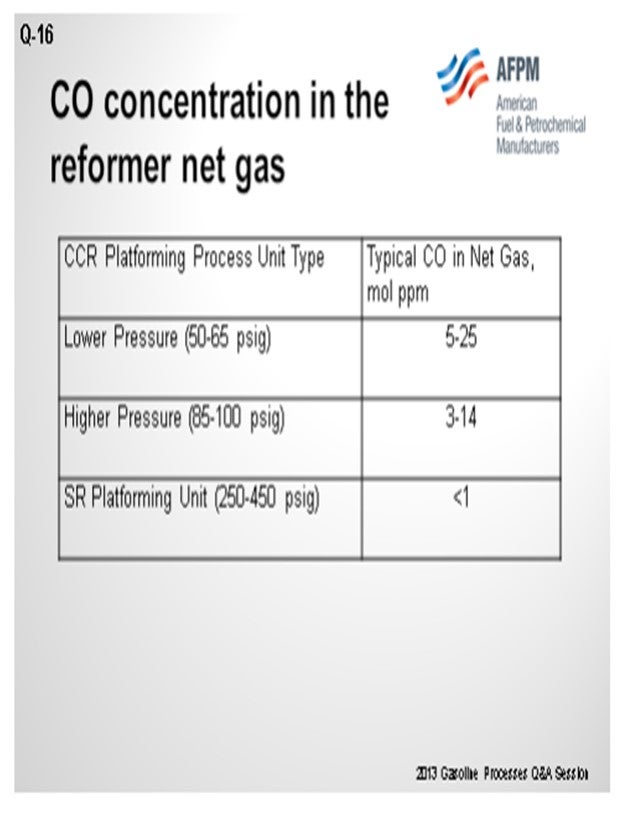
As you can see in this table, we have correlated some different types of operation and ranges of CO levels. As Craig alluded, the numbers vary quite a bit, which can be due to conditions in the unit, as well as analytical capabilities. There seems to be a trend that the lower pressure units generate more CO than higher pressure units.
The laboratory method we recommend using is UOP 603, which is a laboratory method for CO and CO2 and hydrogen. However, a lot of refiners cannot do this method. The gas detection tube route is fairly common. Our point of view is that with the gas detection tubes, if one carbon pre-tube is good, then two is better. So, we usually ask them to use two tubes instead of one to help eliminate the breakthrough of hydrocarbons that can make a false high value for CO.
As Craig said, the issue with chloride and alumina isomerization catalyst is that you will deactivate the catalyst. However, if you are using another type of catalyst, like the Par-Isom catalyst or zeolitic catalyst, the actual suppression you will get will be very dependent on what temperatures you are running. As you approach the 400°F temperature, you tend to methanize the CO in the first part of the bed. So CO tends to have less of an effect on the metal function of the isomerization catalyst and becomes more of an issue of activity suppression due to the water on the acid sites. The same would apply if you had a saturation unit with platinum catalyst. It would also behave in a similar manner to these higher temperature isomerization units.
R.K. (RICK) GRUBB (Chevron Products Company)
Another aspect needs to be mentioned for the lower pressure reforming units. You have to take into consideration your nickel carbonyl formation when you shut down a hydroprocessing unit that is using the reformer hydrogen. You may have to either swap the hydrogen source or think of another shutdown procedure that will ensure no nickel carbonyl formation.
CRAIG MELDRUM (Phillips 66)
CO is detrimental to downstream hydrogen using units for three principal reasons:
1) CO will methanate in HDS (hydrodesulfurization) units consuming hydrogen, which will take away catalyst activity.
2) Much of the CO will methanate in isomerization units, forming water that will deactivate the isomerization unit catalyst.
3) The non-methanated CO in the isomerization unit will poison the metal function of platinum on the catalyst.
Note: The UOP suggested CO limit on isomerization unit hydrogen makeup gas is 1 ppm (10 ppm for CO + CO2). UOP reports that CO levels greater than 6 ppm will not allow the isomerization unit catalyst to meet its cycle life guarantee.
The common steps to minimize CO formation in the reformer are:
• Minimize moisture in the system (feed water control and good regeneration drying),
• Minimize the last reactor temperature,
• Maximize the H2/HC ratio, and
• Minimize catalyst circulation rate in a CCR.
PATRICK BULLEN (UOP LLC, A Honeywell Company)
The CO concentration in reforming unit net gas can be impacted by a number of factors: system pressure, temperature, and moisture in the recycle gas, as well as the H2/HC (hydrogen/hydrocarbon) of the operation. Operating pressure has the most significant impact on CO production in a reforming unit. CO formation in reforming operation is produced via steam reforming of hydrocarbons:
H2O + CH4 ↔ CO +3H2
Thermodynamically, this CO formation reaction is more favorable at lower pressures. CO production is inversely proportional to the pressure squared. As such, a semi-regeneration reforming unit, being significantly higher pressure than typical continuous reforming units, will tend to produce less CO than a typical continuous reforming operation. Likewise, the lower pressure high severity reforming unit operation is more favorable for CO productions.
Commercial reforming net gas CO data from CCR Platforming™ process units can range from 1 to 40 mol ppm. The table below indicates typical ranges for various unit types. Typically, CO levels in the semi-regeneration reforming units are at trace ppm levels due to the high pressure and low-moisture range operation.
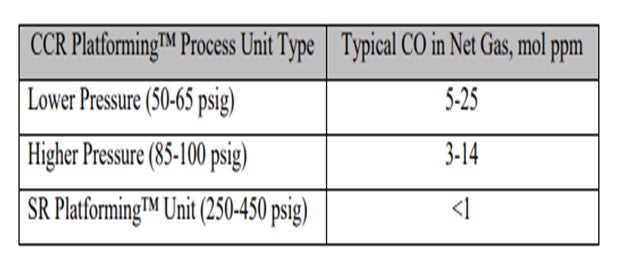
For testing of CO in reformer net gas, UOP recommends method UOP 603 for trace CO and CO2 in hydrogen. For CO in light gaseous hydrocarbons, analysis by GC is recommended. Analysis by gas detector tubes can also be considered for measuring CO at elevated levels when used with several carbon adsorbing pre-tubes.
CO in reforming net gas can have an impact on downstream users that may be sensitive to CO or H2O that may be formed due to the reverse steam reforming reaction, also known as the methanation reaction. In the case of a Butamer™ and Penex™ catalyst, water is a permanent deactivator. A typical rule of thumb is that 1 pound of H2O kills 62 pounds of Butamer™ and Penex™ catalyst.
For other types of catalysts, such as Par-isom™ Process and zeolitic isomerization catalysts that operate at higher temperature, the water generated from methanation of CO is a temporary activity suppressant. Platinum-based BenSat™ catalyst behaves similarly to these isomerization catalysts.
GARY HAWKINS (Emerson Process Management)
With respect to the second part of the question, the carbon monoxide content, as well as other components in the net gas of a naphtha reforming unit, can be measured with a variety of measurement principles depending upon the accuracy and reliability required, other species present that may interfere with a particular technology, and the expected range of concentration of carbon monoxide. These same comments apply to measuring other refinery gases, such as the net hydrogen and PSA (pressure swing adsorption) tail gas from steam reforming units for hydrogen production.
Year
2013
Process
Question 98: What is your experience with the use of ammonia or steam in the FCC flue gas line in order to improve the operation of the ESP? Please comment on system configuration and operational issues
PIMENTEL (CITGO Petroleum Corporation)
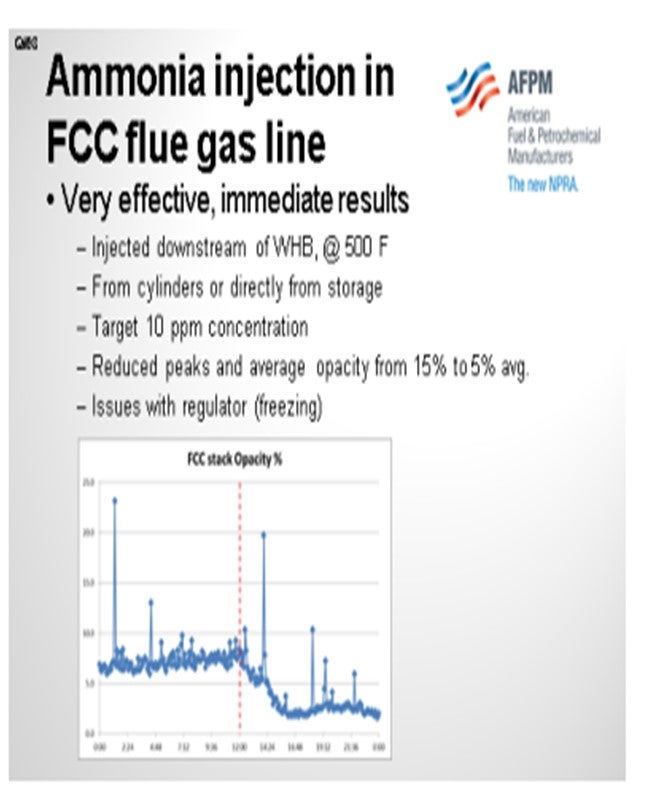
We have extensive experience with the use of ammonia in the FCC flue gas line in order to improve the conductivity of the particles and improve the operation of the ESP. We inject ammonia at the target level of 10 parts per million or less. It is very effective at that concentration and has helped us reduce our baseline opacity level from about 15% to less than 10% or 5%. It also reduced the peaks. The peaks that you see in the chart are related to soot blower operations of the waste heat boiler. Thanks to the ammonia, we now operate the soot blowers more often without the fear of violating the opacity limit. So it is also an energy-saving project.
In our unit, the ammonia is injected directly downstream of the waste heat boiler from cylinders or from a storage tank located outside of the FCC battery limits. The only issue I can recall with this operation has been the loss of ammonia flow due to the regulators plugging with ice. If the ammonia is not completely dry, it will freeze in your regulator. That is easy to fix by putting some steam tracing in the lines. The chart shows typical performance before and after starting the injection of ammonia. As you can see in the chart, it helps us operate the soot blowers more often.
BROOKS (BP Refining)
As Sergio mentioned, the common use for putting in steam and ammonia is to reduce the resistivity of the particulate so it can be picked up easier in the ESP. We do not have a lot of experience with steam helping our ESP operations, apart from the example I mentioned about using steam on start-up before meeting temperatures necessary to use ammonia.
We do have quite a bit of experience with ammonia injection. All of our ammonia injection systems are fairly similar because, again, we use the same consultant for the vast majority of our ESPs. We also found that it is key to focus on your ammonia injection system providing good dispersion in the flue gas stack and good vaporization of your ammonia. Typically, all of our systems include heaters for vaporization and metering injection pumps, so we know how much ammonia we are injecting. The systems also include good dispersion nozzles for the flue gas stacks.
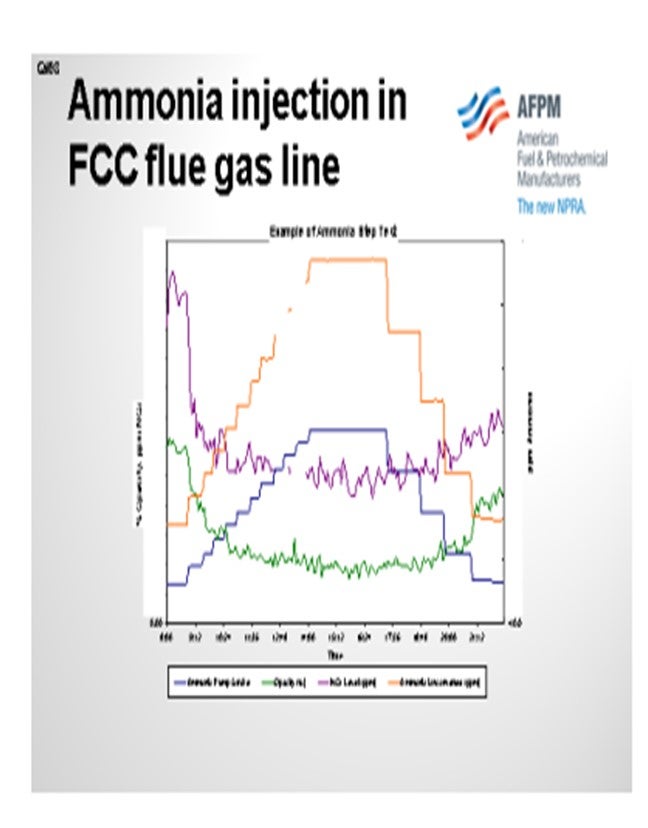
When we use ammonia in our unit, we typically try to optimize it. As shown in the example of an ammonia step test in the slide, the blue and orange lines are essentially the same. They both tell you how much ammonia we are injecting. However, we tend to double-check the meter on the pump stroke versus the actual amount coming in just to make sure we are getting good readings.
You can see that the test includes our stepping up injection rates until we meet a level where we feel like the opacity – the green line – has leveled out. You keep stepping up your ammonia until you believe you have leveled that on opacity, and then you step back down until you feel like your opacity has gone back up. Those are the areas where you would target your ammonia injection rates because you know that is the minimum necessary to maintain your opacity.
I also want to add that I am not sure if other sites have seen instances similar to what we have noticed. Some, but not all, of our sites with ammonia injection into their ESPs also saw a reduction in NOx as a result. It makes sense because you use ammonia in a SNCR (selective non-catalytic reduction) and also in a SCR (selective catalytic reduction) to reduce NOx. However, we do not see it in all of our units.
Those units with high NOx tended to show a good step down and leveling out similar to what we saw with the opacity, which can be seen in the purple line representing NOx. So, you may get an added NOx reduction benefit if you use ammonia on some of your units. We also did the same step test on other units and saw no response to NOx; so, it is not a guarantee.
SCHOEPE (Phillips 66)
I do not have much to add. Halle highlighted all of the points. Phillips 66 has a few installations where we inject ammonia into the ESP. Collection efficiencies were increased by 25% to 50%. It is critical to have good ammonia injection quilts which inject ammonia across the entire duct. Typically, we have not seen any issues with ammonium salt deposition anywhere in the downstream equipment.
MARTIN EVANS (Johnson Matthey Intercat)
To give a contrary comment, I heard a few people talk about the importance of dispersion. We recently had one refiner start ammonia injection and have trouble with the quill. When the quill was removed and the ammonia was injected straight into the nozzle, he got a similar reduction in opacity as he had been getting with the quill. So go figure. It is always the same with the FCC. You can prove something on one unit and then prove the exact opposite in another unit. Another point I want to make is that we have seen that opacity can increase when refiners go to low SOx emissions, typically below 50 ppm and certainly below 20 ppm. This occurs, if you are not using ammonia, because the SO2 (sulfur dioxide) actually acts in the same way as does the ammonia to decrease the resistivity of the catalyst and improve the efficiency of the ESPs. So, when you take out the SO2, you have to replace it with ammonia. Otherwise, you will lose ESP efficiency when you get down to very low SOx levels.
PIMENTEL (CITGO Petroleum Corporation)
We have extensive experience using ammonia in the FCC flue gas line to improve the performance of the ESP. NH3 is a very effective way to improve the conductivity of the flue gas at the levels as low as 10 ppm. In our experience the use of ammonia helped to reduce the flue gas opacity from an average of 15% to less than 5%. Ammonia is injected directly in the flue gas line downstream of the waste heat boiler (at about 500°F) from cylinders or directly from an ammonia storage tank located outside of the unit battery limits. The only operational issue with this system was plugging the regulators with ice, which was solved by steam tracing upstream of the regulator/orifice plate. We do not have experience injecting steam in the flue gas line to improve the operation of the ESP.
BROOKS (BP Refining)
BP does not have a great deal of experience using steam to improve ESP operations. We have one site that uses steam in the ESP during start-up to improve efficiency before the ESP is hot enough to add NH3 injection. This is to prevent possible salt formation that can result from adding NH3 into a cold ESP. The majority of our units use ammonia (NH3) injection successfully to improve ESP collection efficiencies. The purpose of using steam or NH3 injection upstream of the ESP is to condition the particulates by decreasing their resistivity. Decreasing particulate resistivity makes them easier to attract to the walls of the ESP, thus leading to higher collection efficiencies.
As mentioned above, BP uses an industry consultant with a multitude of ESP experience to help guide our ESP operations and optimization efforts. The majority of our NH3 injection systems are similar and follow the consultant’s guidelines, which typically include heaters for vaporization and metering injection pumps. Some sites have basic injection nozzles in the ducts while others have full injection grids. The key considerations for this injection system are around ensuring the injection point provides good dispersion in the flue gas duct and that they NH3 is sufficiently vaporized. Un-vaporized NH3 injection can cause issues with particles remaining on the collecting plates and falling off in chunks or hopper pluggage caused by sticky fines which leads to difficulty evacuating hoppers.
BP has also done a series of NH3 step-tests to optimize NH3 injection. These tests are simple adjustments to NH3 flow rates that are compared to improvements in stack opacity for each step as can be seen in the example graph below. During these tests we have seen that the reduction in opacity with increasing amounts of NH3 injection lines typically lines out at some point, as can be seen in the graph below.
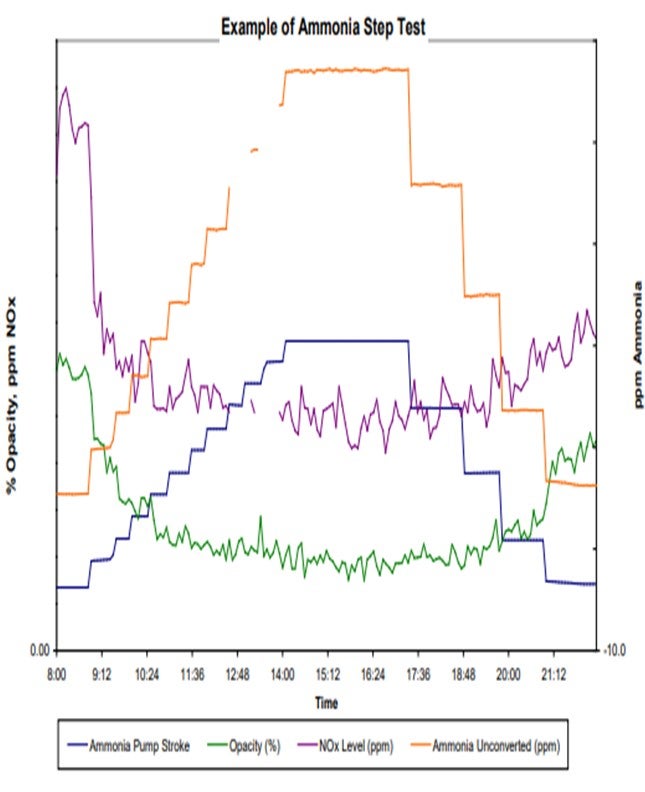
Our experience with NH3 injection has generally been very good at sites with good injection systems. In addition to improvements in opacity with NH3 injections, BP has also seen some reduction in NOx at some of our sites. Generally, we have seen sites with higher base NOx levels see reductions in NOx with NH3 injections and others with lower base NOx levels may not see any change in NOx emissions with NH3 injection.
SCHOEPE (Phillips 66)
Ammonia has been used effectively in a number of refineries to increase electrostatic precipitator (ESP) collection efficiency. Ammonia decreases the resistivity of the catalyst which makes it easier for a catalyst particle to accept a charge. Depending on the ESP design aqueous ammonia injection can increased the ESP collection efficiency by 25% to 50%. A successful installation requires good distribution of the aqueous ammonia. Injection quilts need to be designed to distribute the ammonia equally across the area of the flue gas duct upstream of the ESP and resist catalyst erosion. Deposition of ammonium salts is typically not an issue.
Year
2012
Process
Question 87: The operation of a resid FCC can be challenging as more of its feed is hydrotreated to meet ULSG and ULSD specifications. What changes can be made to improve its operation?
LARSON (KBC Advanced Technologies, Inc.)
For the purpose of this answer, we will look at this as a resid cat cracker that would have a catalyst cooler or be in a two-stage operation. Any increase in feed hydrotreating which increases the portion of hydrotreated feed, or an increase in hydrotreating severity, will obviously improve the feed quality. The slide shows a representative material of a gas oil plus resid and then what it might look like on the basis of a hydrotreated feed. You will notice that by using Watson K or refractive indices, the quality of the feed improves quite substantially. We see that the Concarbon (Conradson carbon) is dropping quite significantly. In those cases, you will have a substantially different coke balance in the unit, and you will see the regenerator fall.
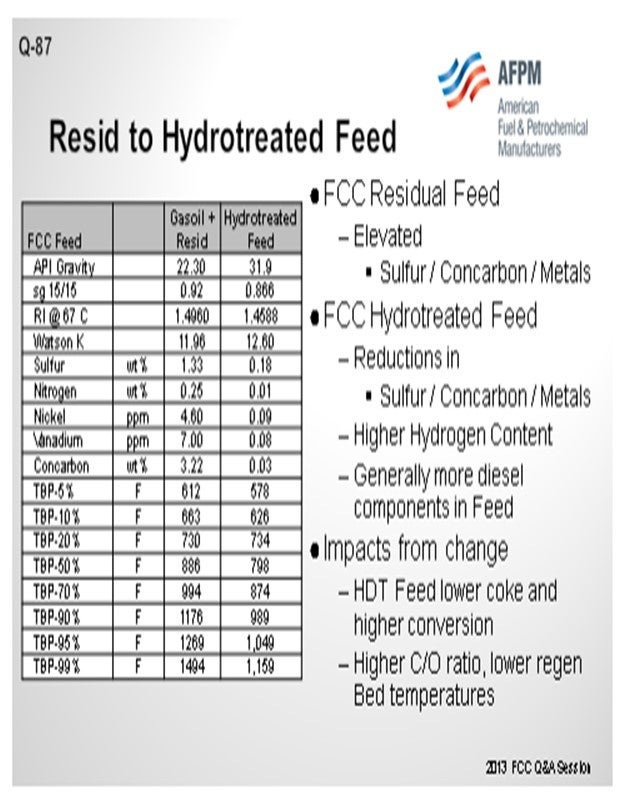
So, what are some of the conditions that you might evaluate to change the operation? One condition is that you will see a much higher cat-to-oil in the unit. Operationally, catalyst circulation itself will go up. The slide valve may need to be increased further. So, then what will be the counter-condition you will need to examine? Can you circulate the catalyst rate that is needed to operate? If you have a two-stage system and a cat cooler, can you keep the cat cooler in operation? Do you just turn it off completely?
Make sure you can get as much trapped diesel out of the unit as possible; because if you distill it in, even if it is only 10%, it will be acting like another cat cooler. With regard to catalyst change, we have a lot of catalyst vendors here. That may be one of the first product you examine in order to maintain the heat balance. You might also consider adding carbon. If you have hydrotreated feed, you can add carbon through the HCO recycle overflash, provided you can maintain the sulfur specifications on your products. You also need to consider the pressure balance to keep the unit operating within targeted range.
In a step change on the existing unit, if you are far enough away from turnaround, you might have to take actions that you would not do in an optimized situation, like reducing the actual steam to the feed nozzle to reduce mixing to allow the addition of carbon. Because it is a situation that exists now and has to be lived with it until the next shutdown, we have clients who have actually pulled steam out of the stripper and let hydrocarbon slip into the regenerator to keep up the profile, which allows them to run the unit. These are non-ideal situations and are done in lieu of mechanical adjustments so you can stay online. Before doing any of the above recommendations, you should consider first a catalyst change.

KOEBEL (Grace Catalysts Technologies)
I want to add one comment about hydrotreated resid. I made a quick query of Grace’s worldwide catalyst database and found some examples of people running hydrotreated resid or partially hydrotreated resid. There were still very, very high levels of metals: much higher than you would consider for a traditional hydrotreated feed operation. Certainly, from a catalyst standpoint, a unit running some hydrotreated resid and a portion of actual resid can still have very high metals; so, you need to make sure you are considering that as part of your solution on the catalyst side as well.
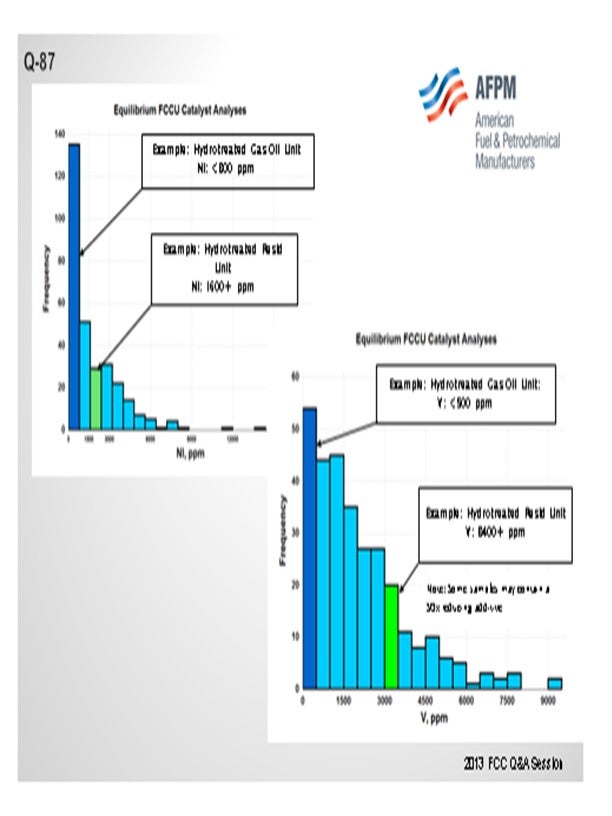
Another phenomenon we see regularly is that the unit running some hydrotreated feed and some resid has a dumbbell-type distribution – a lot of light feed but also some 1300°F plus material – so that type of feed will not behave as if it was blended properly. So, consider the overall distillation of the feed when selecting catalyst as well.
JEFF KOEBEL (Grace Catalysts Technologies)
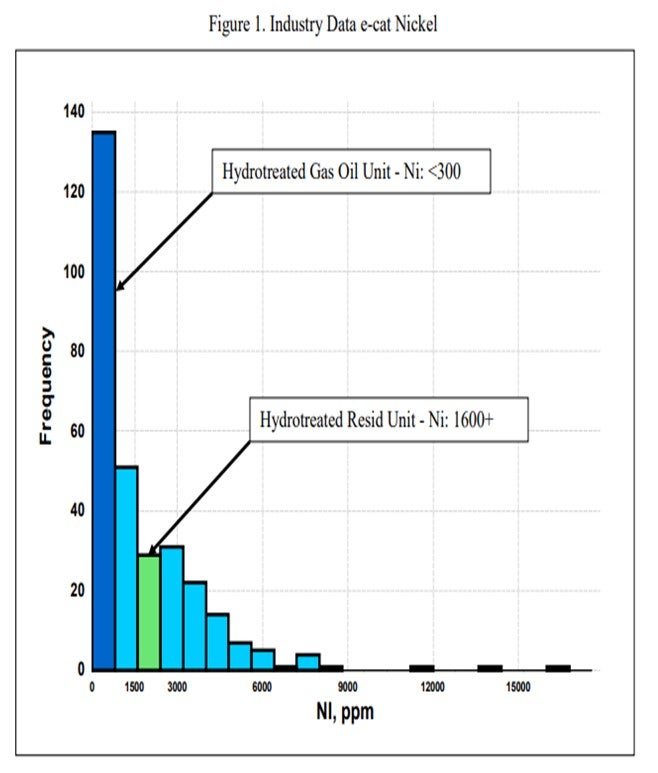
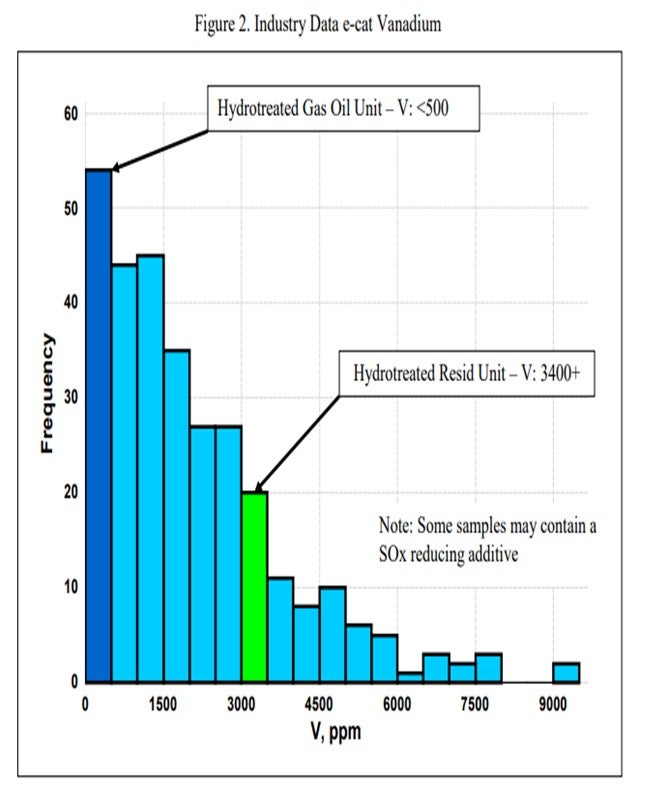
One catalytic challenge related specifically to resid units that process hydrotreated feed is delivering the proper balance of catalyst activity, metals tolerance, and bottoms cracking. Hydrotreated resid, or a mix of resid and some sort of hydrotreated feed, can still contain a significant amount of contaminant metals. One example of a unit processing hydrotreated resid has in excess of 1600 ppm Ni and 3500 ppm V on e-cat, which is much higher than traditional hydrotreated gas oil operations (Figure 1 and Figure 2). With this level of metals contamination, it is important that the catalyst have proper metals tolerance, which one does not normally think of when considering a hydrotreated feed. Having the proper balance of zeolite and matrix activity is also critical to achieve an optimal level of bottoms conversion. In instances where the feed is a mixture of hydrotreated and untreated resid, the resulting feed blend will not behave at all like a feed with its blended API and K factor. Therefore, the capability to optimize the zeolite and matrix balance in the FCC catalyst becomes even more critical.
MEL LARSON (KBC Advanced Technologies, Inc.)
For the purposes of this answer, the designation of resid FCC will be defined by those units with either a catalyst cooler or a two-stage operation. Any increase in feed hydrotreating (either increased hydrotreater severity or an increased portion of the feed being hydrotreated) will improve the feed quality resulting in a lower regenerator temperature. Resid cat crackers, which are designed to burn large amounts of coke, often have problems maintaining sufficiently high regenerator temperature with treated feeds.
When the coking tendency of the feed drops or lowers dramatically, it is a good time to review the crude and vacuum unit operations and any post-hydrotreating fractionation to ensure that the diesel content of the FCC feed is reduced as much as possible since light boiling feed will reduce regenerator temperature. We have found several instances where a minor revamp of the vacuum unit allowed an increase in the vacuum gas oil cutpoint by up to 90°F (or more). This results in more and heavier charges to the FCC, which will increase regenerator temperature.
The normal considerations with lower regenerator temperatures are catalyst circulation issues and regenerator combustion profiles and carbon removal from the catalyst that are very different than the design basis. Therefore, the objective is how to maintain regenerator temperatures that keep the unit within reasonable operating parameters. Operational changes are defined as follows:
• Minimize heat removal via catalyst coolers or quench systems as much as possible. Longer term and depending upon the situation, eliminating regenerator heat removal system could be considered.
• With two-stage regeneration systems, minimize air rate and coke burning in the first stage and shift it to the second stage so a greater percentage of the coke is burned in the total combustion mode. For single-stage regenerators with cat coolers, move toward total combustion with minimum excess oxygen in the flue gas if the unit is in partial combustion.
• Consider a catalyst change. RFCC (resid FCC) units typically use catalyst with low coke selectivity to help minimize regenerator temperatures. Whenever a significant change in feed quality is anticipated, a catalyst evaluation should be conducted to assure that the best catalyst for the new operation is selected. Moving from a low coke-selective catalyst to a high coke-selective catalyst can add 30°C to 50°C to the regenerator temperature.
• Add slurry recycle to the riser.
• Lower stripping steam.
• Lower dispersion steam.
• And lastly, introducing torch oil (in the extreme case) can be considered. The addition of an external fuel source directly to the regenerator (torch oil) has a deleterious effect on the FCC catalyst.
Lower coke yield or lower regenerator temperatures can be an especially severe problem for units with two-stage regenerators. With some of these designs, some coke must be burned in partial combustion; so, there may well be insufficient heat available to run the unit on gas oil or severely hydrotreated resids. This really highlights the need to consider feed flexibility when designing new units as changes in relative crude prices, crude availability, or product specifications (especially sulfur content) can make resid cracking unattractive.
Despite the drawbacks of reducing stripping steam mentioned earlier, we have at least two clients who, after exhausting the other options, have found it economical to do this rather than continue to charge resid to the FCC. In one case, this was a temporary solution used until a catalyst reformulation could be put into effect. In the other, it is still used as a trim variable.
Hardware changes to consider accommodating higher catalyst flux rates would include, but not be limited to:
• Elimination of heat removal system on regenerator,
• Review of standpipe and slide valve sizing, and
• Expansion of capacity given that the air blower is less constrained.
Consider post-treat options that allow a more carbonatious feed.
Year
2013
Process
Question 85: What are the advantages and disadvantages of catalyst mesoporosity [100 to 600 ǻngström (Å) diameter pores] in VGO (vacuum gas oil) operations?
KOEBEL (Grace Catalysts Technologies)
When talking about catalyst mesoporosity, we are referring to the matrix contribution, especially the 100 to 600 ǻngström pores found in the catalyst matrix activity. Catalyst matrix activity is really important, particularly in the Type I cracking depicted here, in the mix zone of the riser where the mix zone can be on the order of 1100°F. Even in VGO types of operations, you can have a significant portion of the total feed that may not vaporize at those conditions. Having effective diffusion is important for pre-cracking these types of feeds so that the zeolite can efficiently crack it further later on up the riser. Certainly, even in VGO operations, we think that avoiding diffusion limitations in the mix zone can be tremendously important.

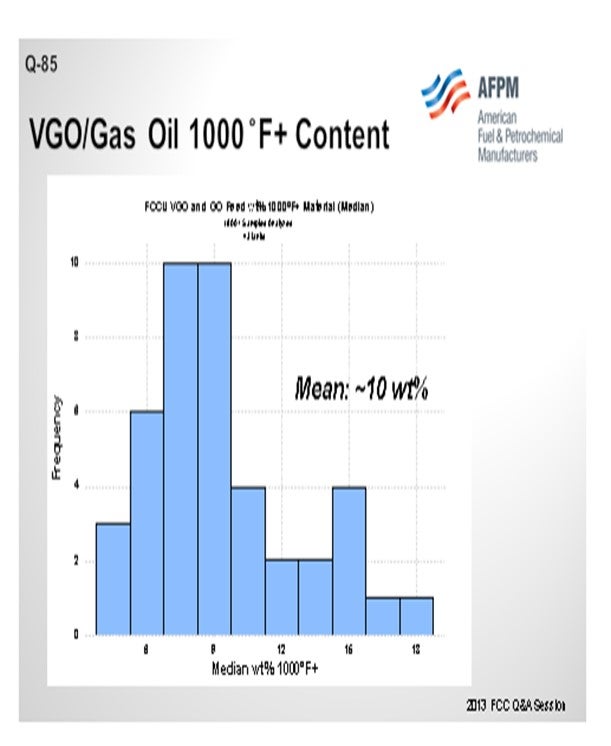
I queried the Grace database on FCC feed. Even in gas oil and VGO operations, the average was that about 10% of the feed boiled over at 1000°F; so clearly this is not just a resid phenomenon. This is a factor in FCC operation that Grace considers to be very important in even the VGO operations.
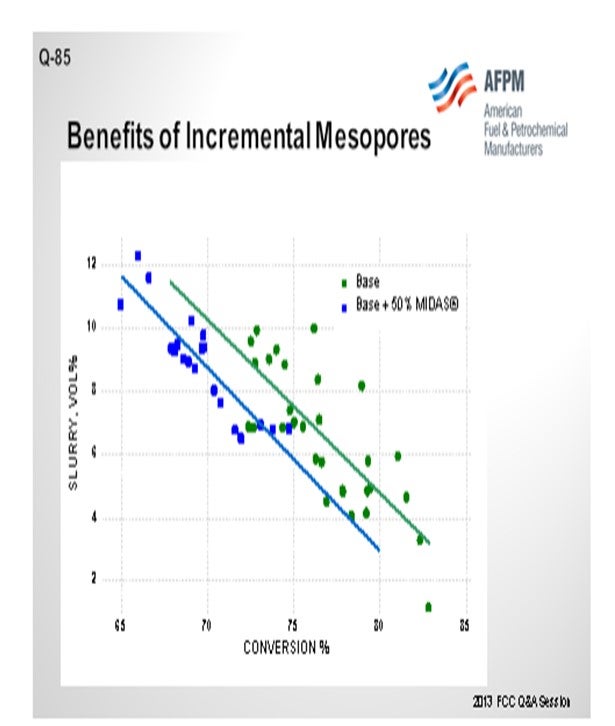
The next slide shows a commercial example of a refinery that was running on a catalyst and implemented 50% of Grace’s MIDAS® catalyst into the blend. MIDAS® is a catalyst for which Grace specifically maximizes the mesoporosity and matrix surface area in the mesopore range. You can see that over a wide range of conversion, the slurry yield was reduced by 2.0% absolute. This is a gas oil operation. Even though this would not be a feed you would consider to be heavy resid, clearly the matrix contribution to the efficiency of the cracking was obvious.
The question also asks about possible disadvantages of mesoporosity. I believe that it might be a disadvantage to have too much mesoporosity in the catalyst when there is a need for high zeolite activity, which is certainly very important, particularly in this Type II cracking where you are going to try and de-alkylate side chains off of naphthenic and aromatic cores. This cracking is very efficiently done by zeolite. If you have a catalyst system that is too biased towards matrix surface area (MSA), you may have a deficit in zeolite activity. So, you certainly want to make sure you have the proper Z/M (zeolite/matrix) ratio on all feeds.
JOE McLEAN (BASF Corporation)
We introduced a catalyst called NaphthaMax® back in about 2000. Really, the only difference between NaphthaMax® and its predecessor-type technologies was that there was a higher porosity version. It has been, by far, our best-selling gas oil catalyst line ever since. We are continuing to refine and introduce newer versions of it. Obviously, I agree with Grace, and certainly with Jeff, that it does make a difference, even in gas oil applications, because it has. We have added over 100 NaphthaMax® users over the past decade or so who could attest to that benefit.
It should not surprise you that we will differ in our opinion a little. I am talking about the zeolite versus the matrix; because with our DMS (dimethyl sulfide) technology, we can put in the mesoporosity. We actually use the external zeolite surface that now gets exposed and available in the larger pores as it functions the way that catalyst is manufactured. So even with the higher porous systems, we are able to get just as high of a zeolite content; and in fact, better zeolite efficiency and activity than what we have with less porous systems generated with the predecessor technologies that we had.
KEN BRUNO (Albemarle Corporation)
Indeed, we agree that mesoporosity is important; but when you talk about diffusion, it extends beyond mesoporosity. In addition to intra-particle diffusion, what is also critical is surface diffusion. To quantify that, Albemarle often talks about the Accessibility Index. Again, mesoporosity is important, but what is more important is the combination of surface and intra-particle diffusion as measured by the Accessibility Index.
PAUL DIDDAMS (Johnson Matthey INTERCAT, Inc.)
Just a quick comment: Do not forget that there are also concentrated additives which contain these kinds of mesoporous cracking sites.
JEFF KOEBEL (Grace Catalysts Technologies)
Effective bottoms cracking involves a three-step bottoms cracking mechanism which was discovered by Zhao1 (Figure 1). The first step is largely dependent on the catalyst matrix and porosity, and that is the effective pre-cracking of large molecules in the feed.2 Since the riser mix zone temperature can be on the order of 1050°F, feed components over 1000°F+ will not readily vaporize. Thus, these molecules need to pre-crack on the catalyst matrix before the catalyst zeolite can further crack them to desired products.
Grace’s FCC feed database contains many examples of units processing VGO and GO that still have a significant portion of the total feed to the unit with a boiling point above 1000°F. In fact, the average 1000°F+ in VGO and GO FCC feed is approximately 10% across our database (Figure 2). Thus, pre-cracking is a real consideration, even in a non-resid operation.
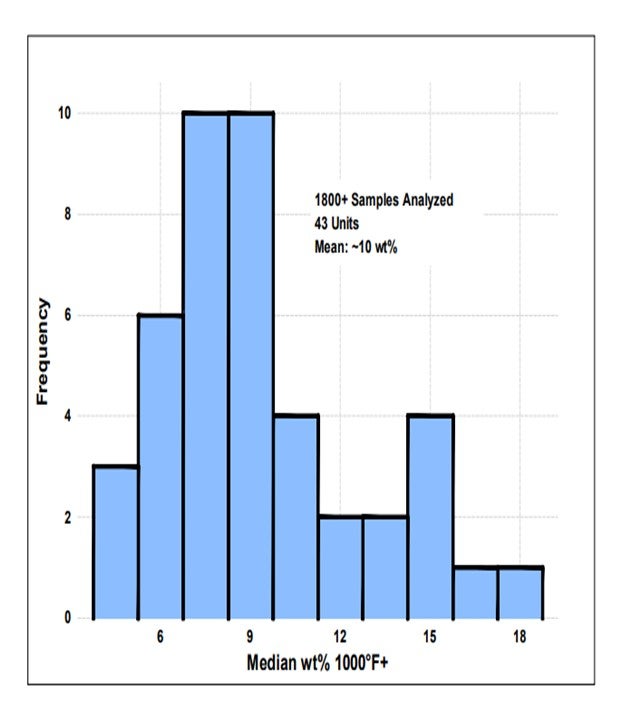
The pore volume of an FCC e-cat (equilibrium catalyst) sample is typically analyzed and reported by each vendor. However, the size distribution of the pores is also a critical component of the effectiveness of the catalyst porosity. Liquid phase diffusion, like one can find in the mix zone of the riser, is typically 2 to 3 orders of magnitude slower than gas phase diffusion, especially if the pores are small (less than 100 Å).4 High mesoporosity in the 100 Å to 600 Å range mentioned in the question is critical for achieving effective pre-cracking of heavy molecules without diffusion limitations. Excess small pores less than 100Å will lead to high coke and gas make, while an abundance of large pores greater than 1000 Å will have ineffective bottoms cracking and perhaps poor attrition characteristics.
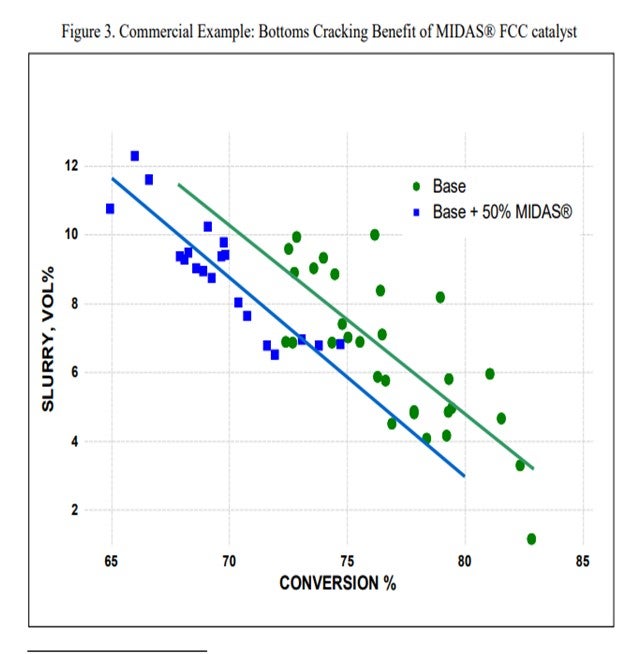
Grace’s MIDAS® catalyst family is specifically designed to provide higher matrix surface area (MSA) with an optimal pore size distribution that provides excellent mesoporosity. Grace has many commercial examples of GO and VGO operations that have benefited from the inclusion of some portion of MIDAS® in the fresh catalyst mix. This commercial example shows that inclusion of MIDAS® into a catalyst system for a VGO application improved bottoms conversion by 2% absolute (Figure 3).
The question also asks about possible disadvantages of mesoporosity. As was mentioned previously, there are multiple steps to effective bottoms cracking. Another step in the conversion of bottoms is dealkylation of side chains from aromatic and naphthenic cores (Figure 1). This is a step that is most efficiently catalyzed by zeolite activity. If a catalyst formulation is too heavily biased towards MSA in an effort to achieve high porosity, it is possible that the formulation may not have adequate ZSA to efficiently drive this type of bottoms conversion. The proper balance between matrix and zeolite is heavily dependent on feed properties and unit yield objectives. Optimization of matrix porosity and zeolite activity is critical to achieve the most effective bottoms cracking, even in light feed applications.
CAREL POUWELS (Albemarle Corporation)
Mesoporosity is typically considered an important feature for enhanced bottoms conversion. This is not only valid for resid applications, but also holds for VGO operations. The effect, however, depends on many aspects, such as the type of VGO, severity of operation, and the base catalyst in use: the more crackable material present in the slurry fraction, the more potential impact by additional mesoporosity. This is particularly valid when the additional mesoporosity is applied within the same catalyst technology. Changing catalyst technology with additional mesoporosity will not necessarily yield the desired effect as the activity, and selectivity of the mesopores between the catalyst technologies can be different. Another aspect that plays a role is catalyst accessibility, which is often confused with catalyst porosity. Poorly designed catalysts can measure high mesoporosity but are not effective as constricted pore openings act as bottlenecks and result in a low accessibility. Thus, it is key to design a catalyst with high mesoporosity and high accessibility.
While mesoporosity (and accessibility) plays an important role in bottoms conversion, it also contributes to the preservation of important primary cracking products such as olefins in the gasoline range. These are important components for high gasoline RON (research octane number) and also precursors for the production of propylene.
Another potential benefit of catalysts with reduced mass transfer limitations in VGO applications becomes evident in units with advanced riser termination devices (RTDs) and short contact times. Under these circumstances, reactants can travel faster to the active sites, and products can egress from the pores before the catalyst is separated from the hydrocarbons in the RTDs, resulting in higher conversion and reduced rate of secondary reactions (everything else constant, compared to a catalyst with inferior pore architecture).
Albemarle offers a wide suite of products for different VGO applications with a varying range in mesoporosity and accessibility, such as AMBER™ for lowest slurry, GO-ULTRA™ for maximum gasoline, ACTION® for superior gasoline octane and alky-feed, and AFX™ for record high propylene.
Year
2013
Process
Question 34: When processing cracked naphtha, what is done to ensure that polymerization of the diolefins/olefins will not result in pressure drop problems in a reactor or upstream equipment?
GATES (Motiva Enterprises LLC)
Again, we are trying to prevent the polymerization of the olefin/diolefin. The primary concern is trying to prevent contact with oxygen because that will ultimately lead to gum formation. So the preference would be, if possible, to feed this hot to all the downstream units and avoid intermediate storage. If you will have to put it into tankage, the preference would be to use an internal floating roof tank and add some type of chemical stabilization. So typically, some kind of oxygen scavenger would be put in there. The length of time that material will be staying in tankage may dictate the level of treatment that will be done.
The other concern is that if this material contains diolefin/olefins, as it goes into your feed/effluent exchangers, prior to entering the reactors, it could potentially foul those exchangers. Often, you want to add soak hydrogen or hydrogen upstream of that exchanger. It may need to be considerably upstream to ensure that as the material is going into the exchanger and getting hot, there will be some hydrogen present to prevent that gum formation.

If you are actually going to try and treat cracked naphtha, you will typically want a separate diolefin reactor that will be run at lower temperatures to avoid the polymerization. You will use catalytic hydrogen to eliminate the diolefin and operate it in a regime such that you will not saturate too many of the olefins. You are ultimately trying to put the naphtha into gasoline, so you want to minimize your octane loss for the gasoline pool. As far as the catalyst, look for a catalyst with sufficient surface area and large enough pore volume – going back to Sal’s comment – to manage any silicon or arsenic that might be coming with that cracked naphtha.
WATKINS [Advanced Refining Technologies (ART)]
Two of the reasons we are worried about olefins and cracked naphtha in our naphtha hydrotreater are that they are a large consumer of hydrogen and also because the reactions occur quickly. Once the reaction with olefins starts and we have enough temperature, it is hard to stop. We do not want excess temperature to coke up at the top of our hydrotreater. Like David said, with coker feeds, we would like to see them brought in straight from the coker while still hot. They should not be sent to tankage; and if they are, you should blanket the tank somehow and keep it protected from oxygen.
The top of the hydrotreater is where we are worried about excess exotherm. We want to spread out that heat and do the reaction somewhat slowly, if possible. We do not want to have it occur all at once. Generally, we will recommend a graded bed to mitigate that pressure drop and start olefin saturation to avoid worrying about anything else like HDS and HDN or aromatics. In that case, we will be using a low-metals guard material at the top of our hydrotreaters, which also gives us the ability to pick up silicon.

If your system has a high amount of diolefins, again, a dedicated diolefin reactor is important. At a much lower temperature, you can actually control just the diolefin reaction and avoid doing everything all at once by either controlling how much hydrogen goes to the hydrotreater or by keeping the temperature fairly low.
In the top of the main reactor of a coker naphtha unit, again, low metals, a guard material, is recommended. We generally then recommend avoiding the placement of very high-metals, high-active catalysts at the top, if at all possible, so we can try and do the reactions one at the time.

MUKESH PATEL (Reliance Industries Ltd.)
When we have to enable storage of naphtha, if necessary, which is the parameter we should monitor or what type of analysis should be carried out to determine if any gum formation has happened?
GATES (Motiva Enterprises LLC)
Off the top of my head, I do not know the ASTM (American Society for Testing and Materials) methods for that, but there are gums and then there are potential gums a
We run diolefin saturation tests, and I will put the ASTM method in my Answer Book response. We typically shoot for 60 to 80% diolefin saturation.
SHARPE (Flint Hills Resources, LP)
We run diolefin saturation tests, and I will put the ASTM method in my Answer Book response. We typically shoot for 60 to 80% diolefin saturation.
BRIAN WATKINS [Advanced Refining Technologies (ART)]
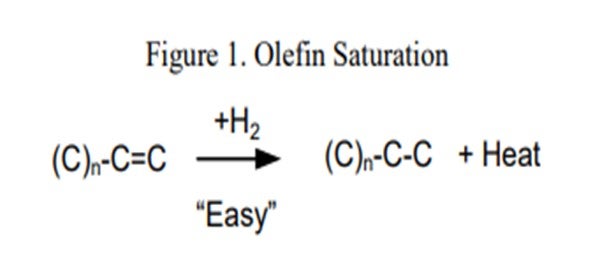
Processing coker naphtha can have several undesirable effects on the performance of the hydrotreater and the catalyst if the system was not properly designed to handle it. In general, coker stocks have a higher level of olefins present from the coking process. Once in the hydrotreater, these olefins will quickly get saturated (Figure 1), resulting in high hydrogen consumption and generation of a lot of heat. As a general rule of thumb, one mole of hydrogen is required per mole of carbon-carbon double bonds, or between five and 10 times the Bromine number reduction in standard cubic feet per barrel (scfb) of hydrogen. This additional heat [130 Btu/scf (British thermal unit per standard cubic foot) to 160 Btu/scf hydrogen consumed], if not managed properly, will initiate additional reactions, quickly creating a very high temperature rise. The high temperatures can accelerate coking and can lead to olefin polymerization, resulting in a dramatic increase in pressure drop. This can set an upper limit on how much coker naphtha can be processed based on the need to limit the heat rise or hydrogen consumption.
A system that is properly size and activity graded is extremely important when processing coker naphtha. ART recommends utilizing a grading system to help mitigate pressure drop buildup. A large inert hold down ring (GSK-19) with a very high void fraction used for trapping large particulates is placed at the top of the catalyst bed. A smaller diameter macroporous ring (GSK-9) that traps iron, as well as other fine particulates, is typically used in the next layer. After that, two types of smaller rings are used as active grading. These materials have a low level of active metals which help begin olefin saturation reactions, as well as provide additional particulate space at the top part of the catalyst bed. Avoiding the use of any highly active catalyst at this point is also recommended. Below the grading system, it is recommended to use a layer of larger size (1/10” or 1/12”) catalyst which provides activity for olefin saturation and additional void space for pressure drop mitigation. This layer is often a catalyst that is suitable for trapping silicon (and arsenic), which is another concern when processing coker naphtha.
An additional recommended practice to help prevent fouling and pressure drop buildup in coker naphtha units is to avoid contact of the coker feedstock with oxygen. It is preferred to bring the feed to the processing unit directly from the coker and avoid use of tankage. If this is not an option, then the alternative is to use a floating roof tank and purge the system to keep a nitrogen-blanket over the feed in order to keep oxygen out. Use of a diolefin treating reactor can also be considered. A diolefin reactor is operated at a much lower temperature in order to selectively catalyze the diolefin saturation reaction and avoid any excess heat generation from sulfur and nitrogen removal.
RAJESH SIVADSAN (UOP LLC, A Honeywell Company)
When processing cracked naphtha, plugging in feed exchangers and heaters may be caused by coking or polymer formation. Polymer formation can be caused by oxygen in the feed with cracked feedstocks. It is preferred to have cracked feeds fed to the hydroprocessing units directly from the upstream unit. Cracked feedstocks that do not come directly from the upstream unit should be stored in nitrogen-blanketed tankage. For units that process straight-run feeds from tankage, as well as cracked feeds, this straight-run feed should also be stored in nitrogen-blanketed tanks as this feed could provide the source of oxygen for polymer formation.
Processing these streams by themselves usually requires a two-reactor system with the first reactor operating at lower temperature to saturate the diolefins and a small portion of the olefins. The second reactor completes the olefin saturation and removes the sulfur and the nitrogen.
DENNIS HAYNES (Nalco Champion Energy Services)
Some issues may arise regarding polymerization. Polymerization in the feed stream may lead to fouling of heat transfer equipment and plugging of the reactor bed. A first step would be to minimize any potential for oxygen contamination as oxygen would act to accelerate fouling reactions. In addition, consideration may be given regarding the implementation of additive chemistries to inhibit polymerization rate (antioxidants) and minimize the potential for foulant (dispersants).
CHRIS CLAESEN (Nalco Champion Energy Services)
There are special low temperature reactor designs utilized to saturate the diolefins before they can thermally polymerize. Chemical programs with antipolymerants and dispersants can also help control polymerization and ∆P (delta P; pressure differential) increase. Reactor bed cleaning programs have been used successfully to reduce reactor ∆P caused by fouling with polymerized olefins and corrosion products.
RAJ PATEL (Haldor Topsøe, Inc.)
Designing a unit for processing coker naphtha can be challenging. The challenge comes from the silica in the feed which can poison the catalyst, high nitrogen in the feed which may be difficult to remove in low pressure units, and high quantity of olefin which can cause large temperature rises in the reactor, as well as very reactive molecules which are the conjugated diolefins. In the presence of small amounts of oxygen, or at elevated temperatures above 450°F, these molecules will radially polymerize to form gum that can foul exchangers or reactors causing poor heat transfer, as well as high rector pressure drop. If the feed contains a significant quantity of coker naphtha, then these diolefins must be removed at low temperatures to prevent gum formation.
The coker naphtha should preferentially be sent directly from the coking unit to the hydrotreater to prevent oxygen contamination. Even straight-run stock, which may be part of the feed component, must be prevented from contacting oxygen by storing the feed in a nitrogen-blanketed storage tank.
Even with strict adherence to avoiding feed contact with oxygen, the diolefins in the coker naphtha can polymerize at elevated temperatures. A dedicated diolefins reactor operating in the range of 300°F to 450°F will ensure that these highly reactive species are removed from the feed before polymerization can take place. Once the diolefins are removed from the feed, then the feed can be heated to the required temperature for silicon and/or sulfur/nitrogen removal. Topsøe has designed a large number of coker naphtha units with dedicated diolefin saturation units to mitigate the polymerization issues.
MIKE ROGERS (Criterion Catalysts & Technologies)
Hydrotreatment of cracked naphtha derived from FCCU, coker, or thermal cracking is a common refinery requirement. In some cases, refineries process these naphthas in a downstream catalytic reformer; while in others, hydrotreatment is necessary to meet gasoline blending sulfur requirements. Treatment of cracked naphtha can pose special challenges due to the presence of diolefins that are produced by the cracking reactions.
Diolefins in cracked naphtha streams from the FCCU and the coker will polymerize when heated, producing gums that foul the pre-heat equipment and main hydrotreater catalyst. To prevent polymerization, these naphtha streams are treated in a diolefin reactor operating at low but sufficient temperature to hydrogenate the diolefins. Typically, the diolefin reactor operates in a down-flow bubble phase with an LHSV of 2 hr-1 to 5 hr-1 and within temperatures between 180°C and 220°C (350°F to 420°F).
The effectiveness of diolefin saturation and protection against polymerization is monitored by measurement of the diene number in the reactor effluent. Typically, method UOP 326-08 (Diene Value by Maleic Anhydride Addition Reaction) is used to generate a MAV (maleic anhydride value) number representing approximately double the diene content in weight percent. Conversion of MAV in the diolefin reactor is in the range of 90 to 95%, and a target MAV number of less than 1 in the product stream from the diolefin reactor typically ensures protection against polymerization.
Catalysts loaded into a diolefin reactor should have a moderate but stable activity to selectively convert diolefins at low temperatures. For units processing coker naphtha containing silicon, a large surface area and high pore volume provide resistance to poisoning. Lastly, for units processing FCCU naphthas, catalysts with a low hydrogenation activity are favorable to minimize olefin saturation and octane loss.
Year
2013
Process
Question 81: How do CO (carbon monoxide) and NOx (nitrogen oxide) emissions change when you operate at low regenerator temperatures? What can be done to mitigate any increases?
BULL (Valero Energy Corporation)
I will initially address this question from a CO standpoint and then discuss the NOx. CO emissions typically increase if the regenerator falls below a certain temperature threshold. That temperature threshold will vary based on your regenerator configuration and definitely on the type of air distribution. In general, well-mixed regenerators with longer residence times can be run at significantly lower temperatures before CO emissions increase, compared to a poorly mixed regenerator with lower residence time.

In the absence of a CO combustion promoter, large variations in the CO2 (carbon dioxide)-to-CO ratios are observed. At the catalyst surface, it is believed that that CO2-to-CO ratio is an intrinsic function of the temperature at the burning site. However, the CO exiting the burning site may be further oxidized to CO2 at a rate dependent on the temperature, amount of CO and O2, water partial pressure, and active metals on the catalyst.
To mitigate CO emissions at low regenerator temperature, we add a non-platinum CO promoter to limit the NOx. Other solutions include increasing 1) your delta coke, 2) the pressure in the regenerator, or 3) the mixing by modifying the regenerator. The amount of NOx emitted from the regenerator is highly dependent upon several variables such as operation regenerator style, excess O2, promoter concentration co-distribution, feed nitrogen, and dense bed temperatures. So, there are many variables that determine the overall NOx equation. NOx formation chemistry inside the regenerator is difficult to pin down. Studies have shown that about half of the feed nitrogen becomes coke on the catalyst, but only a small portion of that actually becomes NOx. It is also believed that NOx emissions are limited by the reaction of CO plus NO (nitric oxide) to form N2 (nitrogen gas) and CO2. Thus, when you have platinum CO promoters added to the unit, CO is converted to CO2 so quickly that there is less CO available for that reaction to occur.
We do have an experience of the regenerator being in true countercurrent flow. With platinum promoter in the unit, it is able to run NOx levels that are very low; but, I will say that is a rarity. In our system, if there is normally a platinum promoter, then you will see elevated NOx levels. In partial-burn operation, there is always CO available to react with the NOx; so emissions are lower. Good coke and air distribution is important to ensure that the concentration of CO, O2, and NOx are evenly distributed throughout the regenerator. The results have been better and more efficient control of the burn zone so that oxygen is used more efficiently and the regenerator can be operated at lower levels of excess oxygen to completely burn the coke to CO2. Generally, you do not need to use platinum promoter. Finally, uniform residence time distribution minimizes the duration that spent catalyst remains the regenerator.

Due to the interaction between CO, NOx, and the FCC regenerator, I recommend that you actually conduct testing to determine the tipping point where you start going up in CO emissions because there are benefits from lowering the oxygen at that time. Running O2 in excess slightly above that tipping point provides a good NOx baseline of your minimum level on the unit. If that level is still too high, then you have other options which include LoTOx™, WGS+™, CONOx™, and SCR (selective catalytic reduction). We have all of these applications across our system. We have very little experience with NOx reduction additives.
GIM (Technip Stone & Webster)
The low regen temperature used to be beneficial. However, with the advent of tight oil and light feed stock, actually too much of a good thing is now bad. So let us first consider what causes some of these low regen temperature phenomena.
The first cause is a result of better feed stock qualities, such as lower Conradson carbon, lower nitrogen, and metals. Second, lower operating severities, such as lower reactor outlet temperature and lower catalyst activity, will reduce regenerator temperatures. The third cause is catalyst regeneration, including full- versus partial-burn, CO promoters (platinum-based or non-platinum-based), and supplemental oxygen that has been turned off.
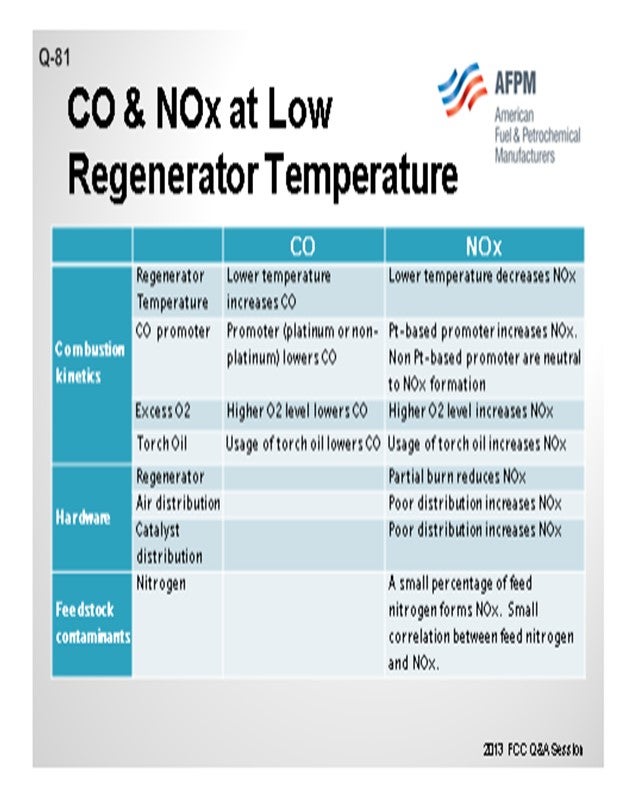
Jeff described many of these aspects, but I thought it would be worthwhile to summarize all of the factors into three main categories. One is, obviously, the combustion kinetics. Second is the hardware of the actual generator design; and third, to a certain aspect, are the feed contaminants such as feed nitrogen. In terms of combustion kinetics, a lot of these items have been discussed multiple times in past panels, so I will just go over the basics and summarize them. The lower regenerator temperature increases the CO but decreases the NOx or NO2; the promoter lowers the CO. But in terms of the distinction between a platinum and non-platinum base, the promoters can be either increased NO2 or NO2 neutral. The higher O2 concentration lowers the CO and increases the NOx. Again, these are some of the factors that you can use to counteract the formation of CO and NO2.
BART de GRAAF (Johnson Matthey INTERCAT, Inc.)
All of the speakers mentioned that the low temperature does affect the CO oxidation and the formation of NOx. The nitrogen kinetics in the regenerator are not affected by the lower temperature. Nitrogen in coke can be converted into nitrogen in NOx, hydrocyanide, and ammonia. As was suggested, non-platinum combustion promoter can be used to steer the oxidation reactions from HCN and NH3 towards N2 instead of NOx. Partial-burn operation is one example where you can see that the CO concentration does affect high NOx. But at the same time, in a partial-burn operation, after the CO boiler, you frequently see a higher NOx level than you would have in a full-burn operation because of the high temperature oxidation in CO boiler.
KEN BRUNO (Albemarle Corporation)
I want to point out that Albemarle has a detailed answer in the Answer Book. But in summary, Albemarle’s non-platinum promoter, ElimiNOx™, provides high CO promotion, excellent afterburn, and minimal impact on NOx emissions.
ROBERT “BOB” LUDOLPH [Shell Global Solutions (US) Inc.]
If you are an oxygen enrichment operator or considering oxygen enrichment, this is your opportunity to raise your bed temperature at the same coke burning rate and shift the CO and NOx distribution within the bed. In turn, the control of NOx and CO emissions could improve.
MICHAEL WARDINSKY (Phillips 66)
We have done a lot of modeling around NOx and CO emissions for additive use. We have seen NOx emissions decrease with higher regen temperatures in almost all of the modeling cases. From our experience, we also know that the non-platinum promoters can increase NOx emission, although not as severely as a platinum-based promoter. We have moved partial-burn units from deep partial-burn to full-burn and observed the NOx emissions decrease substantially. I think it was from the gentleman from INTERCAT who mentioned that in partial-burn, you are releasing ammonia and HCN (hydrogen cyanide) off the regenerator in greater quantities than during full-burn operations. However, these reduced nitrogen species are combusted across the CO boiler to NOx, so your partial-burn units always appear to emit higher NOx emissions than operating in full-burn, even after accounting for thermal NOx generated in the CO boiler.
JEFFREY BULL (Valero Energy Corporation)
CO emissions will increase if the regenerator falls below a certain temperature threshold. That temperature threshold varies based on the regenerator configuration and the type of air distribution. In general, well mixed regenerators with longer residence times can be run at significantly lower temperatures before CO emissions increase compared to a poorly mixed regenerator with lower residence times. In the absence of a CO combustor promoter, large variations in CO2/CO ratios are observed. At the catalyst surface, it is believed that the ratio of CO2/CO is an intrinsic function of the temperature at the burning site (“Arthur's ratio”). However, the CO exiting the burning site may be further oxidized to CO2 at a rate dependent on temperature; CO, O2, and H2O partial pressures; active metals on the catalyst; carbon/oxygen distributions within the fluidized bed; and even the catalyst presence.
To mitigate CO emissions at low regenerator temperatures you can:
• Add non-Pt (platinum) CO promoter to limit NOx,
• Modify your operation to increase your delta coke (catalyst or operational changes),
• Increase pressure in the regenerator, and/or
• Modify the regenerator to increase mixing.
The amount of NOx emitted from the regenerator is highly dependent upon several variables such as mode of operation, regenerator style, excess O2, promoter concentration, coke distribution, feed N2, and dense bed temperature. Studies have shown that about half of the feed N2 goes to coke on catalyst, but only a small portion of the N2 in coke goes to NOx. It is believed that the NOx emission is limited by the reaction of CO + NO to form N2 and CO2. Thus, when Pt CO promoter is added to a unit, CO is converted to CO2 so quickly that there is less CO available to react with the NOx; so NOx emissions increase. We do have experience with a regenerator that is in true countercurrent flow, and the operator is able to run at low NOx levels with platinum promoter in the unit. In a partial-burn operation, there is always CO available to react with the NOx formed; so, emissions are lower. Good coke and air distribution are important so that concentrations of CO, O2, and NOx are evenly distributed throughout the regenerator.
The results of the better, more efficient control of the burn zone in a regenerator are that:
• Oxygen is used more efficiently so that lower levels of excess oxygen are required to completely burn the coke to CO2 while minimizing CO emissions.
• Generally, no Pt combustion promoter is needed (at design rates) to accelerate the burn to completion.
• The uniform residence time distribution permits the time spent in the regenerator to be minimized.
Due to the interaction between CO and NOx in an FCC regenerator, testing should be conducted for your individual regenerator to determine the “tipping point” where a reduction in excess O2 causes CO emissions to increase dramatically. Running at an excess O2 slightly higher than the “tipping point” provides a baseline NOx that you can expect for your unit. If this level is too high, then you will either need to modify your regenerator for better mixing or use an alternate method of NOx reduction like LoTOx™, WGS+™, CONOx™, or an SCR. We have four LoTOx™ applications, one WGS+, one CONOx™, and one SCR in our system. In some cases, a catalyst additive can reduce NOx, but we have limited experience with this application.
STEVE GIM (Technip Stone & Webster)
Why lower regenerator temperatures? Let us first examine some of the factors leading to lower regenerator temperatures: first, better feedstock qualities (such as lower Conradson carbon), lower nitrogen, and metals. Second, lower operating severities, such as lower reactor outlet temperature and lower catalyst activity, will lower regenerator temperatures. Third, catalyst regeneration, including full versus partial-burn, CO promoters (platinum-based or non-platinum-based), and supplemental oxygen turned off are additional options.
Factors Directly Affecting CO and NOx: Many of these directly affect the formation of both CO and NOx in the regenerator. Some of these factors influence the formation of these two species in opposite direction. I have summarized these factors into three categories and their directional changes of the CO and NOx. Many of these items are obvious and have been extensively discussed in the past panels, but I thought it would be a good summary.
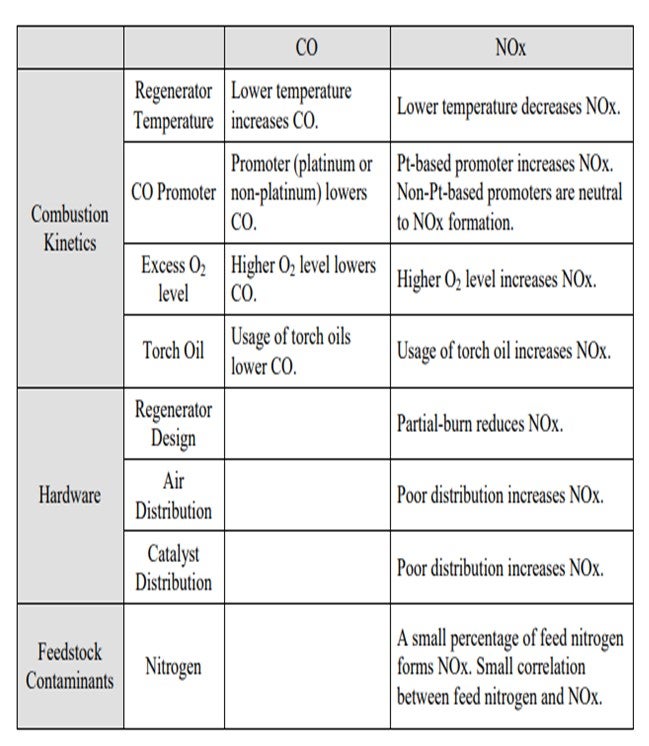
Interactions Between CO and NOx: Reductant-like CO converts NOx to elemental nitrogen in the presence of O2. Lower regenerator temperature can be the direct result of higher CO to CO2 ratio in partial-burn units, which in turn will lower the NOx.
CHRIS STEVES (Norton Engineering)
The impact of low regenerator temperature on CO and NOx emissions can be very unit-specific, depending especially on regenerator design and the distribution of air and coke in the regenerator. In full-burn units, operation at low regenerator temperature will generally result in higher CO emissions and lower NOx emissions. The use of CO promoter can help in reducing CO formation, but use of platinum-based promoters is well documented as increasing NOx emissions. Non-Pt-based promoters are widely used today to allow for control of CO (and afterburning associated with CO) while limiting NOx emissions.
RYAN NICKELL (Albemarle Corporation)
Though not directly stated, this question is most applicable to FCC units concerned with after-burn. Lowering the regenerator temperature can indeed lead to an increase in CO and NO to N2 and CO2. A platinum-based combustion promoter can be used to reduce CO emissions with excellent success. However, NO will increase as intermediates, such as HCN and NH3 (ammonia), are also oxidized by platinum. As a result, many refiners prefer a non-Pt-based promoter such as Albemarle’s ElimiNOx™. This additive provides high CO promotion activity but acts less on the aforementioned intermediates containing nitrogen. ElimiNOx™ provides excellent after-burn control and minimal impact on NOx emissions.
Year
2013
Process