General Session
Speakers
Session Start End
-
Tim Lewer (Shell)
The issue is how to accurately determine the silicon content. Standard Inductively Coupled Plasma (ICP) techniques will give a result that is equal to or greater than the true amount of silicon present depending on if the silicon is in a volatile form (low molecular weight silicones). So, ICP can be used to screen samples and ensure silicon levels are below a desired limit. However, if silicon is detected, it could be a false high number due to nebulizer enhancement effects. Alternatively, a new ASTM method using Monochromatic Wavelength Dispersive X-ray technology may be able to accurately measure even volatile silicon species, but the instrumentation is new to the market and does not have a long track record to verify its applicability. A direct injection nebulizer ICP system will accurately measure silicon including volatile forms but is typically too customized of a system for application in a refinery setting.
Be aware of cross contamination when taking samples for silicon analysis. Some plants have seen false high silicon lab results due to silicone grease used for lubrication or certain silicates used in the manufacture of glass bottles. There has also been cross contamination from puncturing certain silicone sample bottle lids during the sample collection process.
Some plants have established a sample station that takes a small sample every 15 minutes into a compositor that is used for a weekly composite analysis. This method doesn't improve sampling speed but does reduce the frequency and improves accuracy.
Kaspar Vogt (Albemarle)
Inductively Coupled Plasma (ICP) is the technique most used. ASTM D-5708, which is intended for determination of nickel and vanadium in crudes and residual oils, has become the de-facto standard for preparing hydrocarbon samples for elemental analysis by ICP. This is probably the most appropriate method for analyzing the silicon derived from the anti-foam agents added to the feedstock, as opposed to the silicon you might find associated with FCC fines suspended in the sample. The detection limit is around 0.5 ppm. The standard deviation is +/- 0.25 ppm.
During operation, silicon addition is minimized in the coker cycle however the refinery does not want to experience a foam over. Understanding the amount of silicon fed to the hydrotreater is complicated as the anti-foaming agent dosage varies during the coker cycle. More sophisticated continuous sampling systems are available and are capable of sampling hourly or periodically and can create a one or multi coker cycle composite sample.
A spot sample value should not be used to predict the life expectancy of the hydrotreating catalyst; the composite sample will be more useful. We recommend to measure the silicon concentration present on the spent catalyst after the cycle is complete and back calculate the average concentration of silicon in the feed. A properly designed guard catalyst system can significantly lengthen the cycle by catching silicon and other poisons before they reach the main bed high activity catalyst.
Martin Gonzalez (BP)
Spot samples of coker naphtha taken for measuring silicon content can have high variability. To obtain a reliable average, you can use a sampling system that accumulates volume over several hours. X-ray fluorescence technology has developed to where a limit of detection of 0.5 ppm silicon is achievable. Gas Chromatography-Mass Spectroscopy (GC-MS) is also useful to quantify the specific siloxanes formed from decomposition of coker anti-foam.
Gerianne D’Angelo (ART)
Accurately measuring silicon in naphtha streams can be done but it takes a bit of work to get a representative sample of the naphtha. The silicon in the coker naphtha depends on the type and amount of antifoam chemical at the delayer coker unit. Delayed cokers have cycles ranging anywhere between 8 – 24 hours. The coker unit is continually producing a coker naphtha stream during these cycles which is typically being sent from the fractionator straight into the naphtha hydrotreater feed drum. The antifoam chemical is usually not added for the entire coker cycle. This means that the silicon in the naphtha stream will vary with the timing of the coker cycle. In order to get a representative amount of silicon in the coker naphtha stream a composite should be made of hourly samples mixed together for the time of the cycle. For example, for an eight hour cycle eight samples would be mixed and the composite sample analyzed for silicon. To measure the silicon an ICP-MS (Inductively Coupled Plasma Mass Spectrometry) instrument can be used. This instrument/method can measure very low metal concentrations.
Shankar Vaidyanathan (Flour)
Pre-mixing hydrogen with feed ahead of the feed/effluent exchangers improves the velocity and increases the shear stresses. This directionally reduces the fouling tendency; as well as, lowering the film thickness and dependent tube wall temperature in the heat exchanger and the charge heater. Hydrogen also offers the physical benefit of sweeping and helps avoid settling particles.
Design criteria such as flow regime, turbulence, design pressure drop, and velocity are specified in order to minimize fouling. The unit capacity and overall heat balance are often factors in selecting two phase or single phase exchanger. Two phase heat transfer coefficient is better than single phase design hence there is a credit in surface area. In order to fully realize this benefit, a large hydrogen quantity, sometimes up to the unit’s treat gas demand, may have to be sent to the front of the exchanger for pre-mixing with feed oil. A caveat while considering the benefits of pre-mixed two phase designs is not to compare the heat exchanger performance to poorly designed liquid only designs. A high fouling factor originally specified may have inadvertently led to a self-fulfilling prophecy of higher fouling in service. In recent years, even liquid only heat exchangers are increasingly designed with high shear stresses to mitigate fouling.
Process control and metallurgy issues need to be worked out for existing liquid only designs before considering pre-mixing gas. The mixing point should be at an appropriate distance ahead of the feed/effluent exchanger to allow mixing time. A minimum soaker hydrogen addition, just enough to keep the dissolved hydrogen in liquid phase such that the flow could be metered and balanced at heater inlet may be considered. The soaker hydrogen could be in the 50-200 SCFB range depending on the unit pressure, heat balance and hydrogen solubility which is temperature dependent. There is some experience in the industry that shows soaker hydrogen minimizes fouling especially with cracked stocks. Bypassing a portion of the treat gas around the preheat system may also be a preferred way to hydraulically debottleneck existing plants. We do not discriminate between recycle gas and makeup hydrogen since any partial pressure credit and solubility differences for using pure makeup hydrogen are relatively minor.
Martin Gonzalez (BP)
Feed/effluent exchanger fouling in hydrotreaters is often the result of oxygen-induced polymerization of olefins. Chain initiation begins with a free-radical mechanism involving molecular oxygen, disulfides, or pyrrolic nitrogen species. In many cases, it appears that mercaptans can polymerize and olefins content needs to be very high. From our experimentation, we have found that hydrogen may not react chemically to stop such fouling at the relatively mild conditions where such polymerization occurs. The exception may be where diolefins are present in high concentrations such as in hydrotreating of light coker naphtha. However, injection of hydrogen into preheat exchangers is effective for increasing superficial velocity, which helps fluidize particles and flush them out of the exchanger. When feed is on the tube side, the necessary fluidization velocity can be calculated based on particle size distribution analysis. A volume of hydrogen equivalent to make up gas flow rate is likely to be sufficient. Recycle gas or combined treat gas may be used.
Minh Dimas (CITGO) The feed side of our Naphtha Hydrotreater Feed/Effluent exchangers has plenty of H2 but always has fouling issue. We have tried anti-foulant with limited success. In our ULSD unit, the Feed Preheat exchanger (feed/product exchanger) has experienced severe and rapid fouling. Lab analyses confirmed two fouling mechanisms: radical-polymerization and oxygenate-polymerization. The feed side does not have H2. We are going to start using anti-foulant while working on long-term mitigation.
Kaspar Vogt (Albemarle) Antimony (Sb)
The effects of antimony in oil on hydrotreating catalyst have not been directly studied, but we can infer the likely impacts of antimony from a variety of information sources and past experiences.
As background, contaminant metals such as nickel can deposit on the FCC catalyst. This will result in increased dry gas (H2 in particular) and delta coke. Depending on the unit constraints this can lead to lower FCC conversion and lower feed rate. Many refiners use antimony in the FCC riser to passivate the detrimental effects of nickel. Antimony will cover the nickel enriched catalyst surface. Side effects are that the Sb will also cover the CO and NOx promoter metals and make these additives less effective.
Excess antimony mainly accumulates in the FCC slurry. However, antimony can be present in the heavier FCC products which are hydrotreated downstream. If the antimony enriched FCC catalyst fines are entrained into the hydrotreater, they can deposit in the catalyst interstices. This will impact bed pressure drop but not catalyst activity. The bed pressure drop build up can be managed by a guard bed catalyst system of sized and shaped catalysts to increase the void fraction and create more particulates capacity.
By analogy with the FCC experience, we would expect antimony in oil to preferentially coat nickel and cobalt promoter metals on the NiMo and CoMo catalysts. Ultimately, this would completely poison the catalyst. During the buildup of coating/poisoning, the activity will likely see a shift towards direct desulfurization (DDS) vs. indirect/aromatic saturation, thus the hydrogenation-to-hydrogenolysis ratio will change. A given concentration of Sb on catalyst would be expected to have a more severe effect on the catalyst performance in high severity HDS/HDN operations like ULSD and hydrocracker pretreat (HC-PT) service than in lower severity hydroprocessing applications such as NHT and LSD.
We seldom, if ever, detect antimony in the interior of spent hydrotreating catalysts where it would be expected to impact activity.
Furthermore, given its position in the periodic table, we would expect that Sb attacks the catalyst's active (NiMo and CoMo) sites, and that it would be a relatively severe poison, similar to arsenic (As), sodium (Na) and lead (Pb). Therefore, we would expect ≤1.0 wt% Sb would reduce HDN/HDS relative volumetric activity (RVA) by approximately 50% in non-severe applications, and that even lower Sb concentrations could severely reduce catalyst activity for high severity operations like ULSD and HC-PT.
Phosphorous (P)
Phosphorous (P) can come into the hydrotreater feed from:
- crudes
- drilling fluids
- phosphated ZSM
- phosphorous-based corrosion inhibitors and flow improvers
- phosphorous from solid phosphoric acid catalyst
-biofeeds
In catalyst manufacturing, phosphorous added on hydrotreating catalyst acts as a promoter and provides additional acidity to enhance HDN, hydrogenation and cracking reactions. Phosphorous also improves metals dispersion on the catalyst surface.
In one instance, we saw that 3 wt% of phosphorus on the catalyst terminated all the exotherm in bed, although other poisons where also present. Organic phosphorous can penetrate into catalyst pores. In general, our understanding is that the poisoning was similar to sodium where ~1.0 wt% concentration halves the catalyst activity.
In a separate instance, we found SiP coming from a solid phosphoric acid catalyst, used in certain FCC gasoline desulfurization processes with some iron deposited at the external surface of the catalyst extrudate. Some phosphorous and silicon also penetrated the catalyst pores. However after the first 0.1 mm, no contaminant phosphorous and silicon were found on a main bed CoMo catalyst. In this case, a layer of P-Si-Fe had deposited at the pore mouth and restricted the diffusion into the catalyst.
Photos of the outer surface including chemical composition are shown below. They show that Alumina, Molybdenum and Cobalt are homogeneously distributed within the catalyst particle, while phosphorous, silica and iron are located at the outer surface of the particle.

We observed that the Si & P from this process behaves totally differently from Si from anti-foaming agents. There are Si-P particles which cannot penetrate the internal pores of the catalyst and are deposited on the catalyst outer surface. The accumulation of these particles cannot be prevented. Therefore, sooner or later, bridges from particle to particle are formed, thus causing pressure drop buildup.
The bottom line is that the quantitative effects of phosphorous on hydroprocessing catalyst performance and the maximum allowable levels are highly dependent on the source and form of the phosphorous compound. It is also dependent on catalyst properties and the process application.
Martin Gonzalez (BP)
Phosphorus can sometimes be found in crude as alkyl phosphates added to passivate metals or protect against naphthenic acid corrosion. Phosphorus esters in crude may originate from waste oils, or from additives injected into wells to improve recovery. Some of the phosphorus may be in a form that volatilizes into distillate fractions bound for hydrotreaters. We have encountered some Canadian crudes containing phosphorus originating from fracturing fluids used in production. Phosphorus content in light sweet crudes seems to be declining, but it may be becoming more prominent in heavy crudes. There have been reports in the industry of ULSD units suffering catalyst deactivation as result of phosphorus from these crudes. From our experience, at 1 wt% on catalyst, it is reasonable to expect a 15-30% activity loss.
Charles Olsen (ART)
Phosphorous (P) contamination in oil has been traced to frac fluids that are often used in crudes from the Western Canadian Sedimentary Basin. The source is diphosphate esters which are soluble in the crude oil. Refineries that run large percentages of light Western Canadian crude have reported crude column and crude furnace fouling for many years. Improvements made to crude columns to minimize fouling have transitioned the depositing of phosphorous to the downstream hydrotreaters.
Other sources of phosphorous include gasoline slop tanks, imported feeds and lube oil wastes. If phosphorous does manage to make its way into the hydrotreater it will poison the active sites of the catalyst causing a loss in activity. A level of 1 wt% of phosphorous on the catalyst results in roughly 10°F loss in activity. ART recommends that a feed content of < 0.5 wppm be maintained whenever possible as well as the use of feed filters to assist in trapping of phosphorous sediment.
Historically, phosphorous contamination has not been very common, but with the increasing use of opportunity crudes it is being observed more frequently. A recent example is summarized in the table below shows the results of some spent catalyst analysis from a diesel unit. This unit experienced extremely rapid catalyst deactivation shortly after start up. It was so severe that within several months the unit required an unplanned turnaround and fresh catalyst was installed. The spent catalyst analysis indicates the catalysts were exposed to high levels of several poisons including sodium and phosphorous. The contaminants penetrated well into the catalyst bed. The level of contaminants indicates the catalyst in the top half of the bed had lost over 60°F of activity.
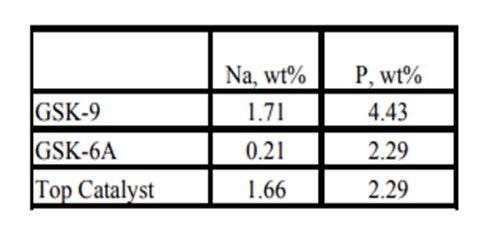
Kaspar Vogt (Albemarle)
A base metal sulfide catalyst will always have less, or hydrogenation compared to a noble metal catalyst. However, in certain situations where deep hydrogenation is not needed, the base metal sulfide catalyst can provide adequate hydrogenation activity. Operation with a base metal catalyst will be between 10 to 20o F higher than a noble metal catalyst, and this will shift the yield towards more thermally cracked lighter products.
Due to the presence of platinum on the noble metal catalyst significant fill cost savings are achieved by loading base metal catalysts.
It would not be recommended to operate a base metal catalyst in a completely sweet service without the presence of H2S. The catalyst risks reduction of the metal sulfides, which will impact the activity and can permanently damage the catalyst.
Effective ways to get around the sulfur stripping of the base metal catalyst in an H2S free environment include spiking by DMDS injection, slipping of some sour gas from the 1st stage over to the 2nd stage, turning off the amine wash, and/or raising the hydrocarbon organic sulfur slip. H2S in the second stage recycle gas is in the range of 15 to 40 ppm to keep the catalyst sulfided.
For a standalone second stage reactor, another issue is corrosion. Since a typical sweet second stage does not have wash water injection, it is important to be careful of metallurgy, especially on the second stage air effluent coolers.
Kaspar Vogt (Albemarle)
In some processes, hydrotreating catalysts are used to treat feedstocks containing very low sulfur (below 20 ppm). These processes can include the following:
1. Gulf HPG process for treating pyrolysis naphtha (second stage).
2. Two step naphtha hydrotreating process in steam reforming (ammonia synthesis process).
3. Treating olefinic fuel gas prior to reforming to make synthesis gas for methanol manufacturing.
4. Wax and certain lube hydrofining operations
It appears that in these processes the catalyst slowly loses its HDS activity. This is due to the transformation of the Molybdenum present as activator, and Nickel/Cobalt present as promoters, from their active sulfide form to the inactive metal state. This process is caused by a hydrogen sulfide partial pressure that is too low (see phase diagrams below).
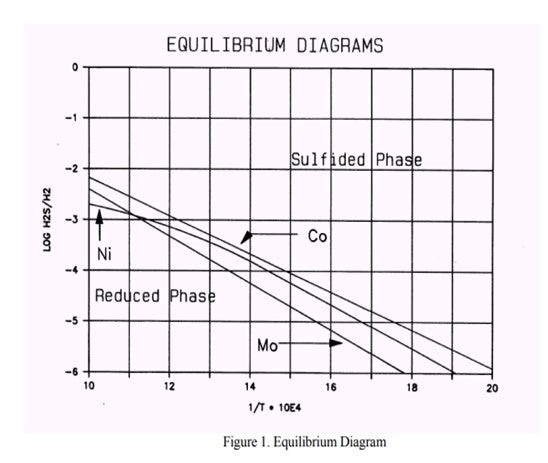
In some cases (usually naphtha units) conditions can arise that contribute to a low H2S partial pressure and could result in metal reduction. Based on the feed properties, operating conditions and product objectives, we can determine whether the unit is or will be operating in a critical operating window. If this is the case, a spiking agent such as DMDS should be added to the feed to boost the H2S partial pressure.
From the phase diagram shown above, we see that the Cobalt is the most critical element. We can begin to move into the Co8S9-phase by decreasing the temperature or increasing the H2S partial pressure by adding sulfur. To move into the "safe" region of the diagram, the Log10 H2S/H2 should be at least -4 at a temperature of 640 deg F.
If the same calculation is done in the reverse order the minimum feed sulfur to avoid metal reduction can be calculated. If we process a 1 ppm sulfur, 70°API naphtha feed at 360 psig at 640 deg F, 80% H2 purity, down to 0.4 ppm sulfur in the product (actual sulfur removal is 0.6 ppm) we conclude that 20 ppm sulfur should be added to avoid catalyst metal reduction. Spiking can be done with a sulfiding agent, e.g., DMDS, DMS.
Minh Dimas (CITGO)
For diesel hydrotreaters, as long as the material being fed to the reactor contains sulfur (i.e., sour feed) and the recycle gas contains 25-50 ppm H2S (by adjusting the amine circulation when necessary), there should not be a need to inject a sulfur-spiking agent into the feed. This ensures a small concentration of H2S at the inlet of the reactor. With reactor temperatures above catalyst activation temperatures, additional H2S is being generated from that point on through the reactor. For us, the sweetest feed is observed when reprocessing off-spec diesel, in which case we may shut down the Recycle H2 Amine Scrubber to preserve the H2S and protect the catalyst.
Tim Lewer (Shell)
In hydrotreating units where the feed sulfur is low and the temperature and H2 partial pressure are low, operation with as low as 20 ppm H2S in the recycle gas has been observed. There is not, however, a universal minimum concentration requirement. The minimum required H2S concentration will vary from unit to unit depending on many factors including, but not limited to: unit feed sulfur concentration, unit pressure, reactor temperature, catalyst type, vent rate, catalyst age, and H2 partial pressure. It is common practice to inject a sulfiding agent such as DMDS to maintain adequate H2S in the recycle gas. In addition, refiners have used sour make up gas streams to provide adequate H2S partial pressure. The guidelines can change based on what catalyst company you talk with, but it all depends on how conservative you want to be. You need to set the H2S low limit to provide a proper buffer zone. It is recommended to discuss H2S concentration requirements for all situations with your catalyst vendor to make sure the catalyst is properly protected against metal reduction.
Gordon Chu (ART)
There is no minimum hydrogen sulfide requirement as long as the feed contains some sulfur as the sulfided catalyst is very resistant to sulfur loss under normal process conditions. We are not aware of any refiners adding sulfur compounds to maintain a minimum H2S concentration during the process cycle.
Tim Lewer (Shell)
On freshly activated catalysts, the surface is relatively clean (free of coke) and therefore is unusually active. This is sometimes referred to as hyperactivity. In order to maximize catalyst stability for good cycle length, it is important that the rate of coke lay down on freshly sulfided catalyst is gradually controlled. Upon completion of metals sulfiding, catalyst hyperactivity exists, but is short lived as feed processing lays initial coke on catalyst. Processing cracked stocks that contain more reactive molecules and coke precursors too early over the hyperactive catalyst can result in operability issues through cracking while accelerating the initial coke lay down on catalyst.
Cracked stocks contain large amounts of aromatics and olefins, which release large amounts of heat when saturated. Aromatics and olefins undergo saturation, but they can also condense or polymerize to form larger molecules, gums, and coke. When cracked stocks are introduced too soon, passing such highly reactive molecules over hyperactive catalysts leads to excessively high reaction rates. The resulting high exotherms aggravate the situation, because reactions like polymerization are faster at higher temperatures. By processing only straight run feeds for the first 72 hours after catalyst activation, the initial coke lay down is gradual for improved long-term stability.
The cracked stock introduction rate varies by unit, but should allow for exotherm stabilization, reestablishment of unit pressure as H2 consumption increases, and coordinated with smaller temperature additions to treat the more reactive feed molecules.
Minh Dimas (CITGO)
I understand “break-in” to be the same as “catalyst conditioning period” – i.e., three days of straight run material only (no cracked stock). It is imperative to adhere to the three full days of break-in (or catalyst conditioning) period following the catalyst activation/sulfiding (which is not included in the three days). After the catalyst is successfully conditioned, the cracked feed can be introduced as quickly as desired by Engineering and Operations (comfort factor) and as manageable by the unit equipment and utilities (i.e., charge heater, hydrogen makeup rate vs. consumption, reactor quench ability, etc.). Since the catalyst conditioning period “quenched” the hyper-active sites of the catalyst, the introduction rate of cracked material after the catalyst break-in / conditioning is negligible.
Kaspar Vogt (Albemarle)
Cracked and heavy feedstocks contain significant quantities of hydrogen deficient molecules known as “coke precursors”. These coke precursors (olefins, poly-nuclear aromatics, asphaltenes, concarbon) tend to polymerize, condense or form coke, given the right set of reaction conditions. Freshly sulfided catalyst, with little or no carbon deposits, is in an ultra-active state. Coke precursors in the feed readily react on the catalyst to produce a molecule with an extremely reactive free radical site. Ideally, this site would react with hydrogen but, because there are so many reactions taking place on the catalyst in its ultra-active state, there can be localized hydrogen deficiency. Without hydrogen readily available to react with the free radical site, the molecule may polymerize or condense with another active molecule, or it may simply deposit on the catalyst surface as coke. All of these outcomes result in blocked pores and/or active sites. Fresh catalyst has hyperactivity due to the absence of coke. Once coke has laid down, the catalyst reaches its design activity, and the coke prevents the agglomeration of metals.
Testing shows the loss of activity between immediate and a three-day waiting period before cracked feed introduction to be 10-15%. We recommend to stepwise increase the amount of cracked stock, which will have a higher exotherm, so that the unit does not incur large temperature swings. It varies per unit depending on the feeds and operating conditions but typically addition of 10% of cracked stocks per two-hour period is good operating practice.
There are commercial offerings available to ex-situ sulfide the catalyst with a special cracked feed protection (CFP) treatment for direct introduction of the cracked feed.
Ben Sim (ART)
Introducing cracked stocks too early after sulfiding will cause noticeable loss in activity. Coke precursor molecules in cracked feeds will have a tendency to form coke over the fresh and highly active sites on the catalyst. By delaying the introduction of cracked stocks for at least 3 days after sulfiding will allow the catalyst activity to be passivated which helps to minimize these effects.
After running for three days on straight run the cracked material should be added to the feed stream gradually. ART typically recommends adding the cracked feed in small increments every shift making sure the reactor exotherm remains under control and within acceptable limits before increasing the cracked feed amount any further.
Martin Gonzalez (BP)
We have used a terpene-based product to help free hydrotreater reactor circuits of hydrocarbon in preparation for catalyst change-out. The product was effective for achieving a low level of combustibles in the reactor and in all vessels of the reactor circuit. However, in one instance, the test for entry indicated high benzene levels, and further nitrogen purging was required. As a result, the benefit of a quicker shut-down was not fully realized. In our trials, the greatest gains in shut-down duration resulted from the review and optimization of cool-down procedure that took place in preparation for the injection, rather than from injection of the chemical itself. Also, note that chemical may heat up the catalyst upon application, due to a relatively high heat of adsorption. Such a warmup may extend cool-down time beyond what was anticipated.
Tim Lewer (Shell)
Some plants have used additives during the hydrotreater reactor cool down period in order to remove hydrocarbon, especially benzene. These additives have been used with varied degrees of success. To this date, data has been inconclusive as to whether or not the chemical additive speeds up reactor decontamination versus conducting a proper hot hydrogen strip.
Injection of chemical additive requires several extra considerations in addition to the expense of the chemical:
• Waste disposal – Chemical suppliers may claim that their product is safe for refinery re-run systems, but most plants will be hesitant to re-run through the crude unit due to concerns, for example crude column overhead corrosion.
• Piping for injection – Temporary piping needs to be installed in order to inject chemical to the desired locations. This creates additional expense and maintenance workload during unit shutdown. Also, many plants may not allow connection to the process until the unit is down to a low enough pressure.
• Hold points – Many chemical additives need hold times at certain temperatures per the manufacturer to guarantee hydrocarbon removal. This will add time to your cool down.
Kaspar Vogt (Albemarle)
We have experience with dumping, screening and reloading hydrotreating and hydrocracking catalyst. Here are two typical times this occurs:
1. After a full cycle – In these cases spent catalyst has been reused, without regeneration or reactivation, in a lower severity application for which the remaining activity is sufficient.
2. Early in the cycle – If at the start of the cycle the pressure drop is very high (due to, for example, broken inert balls or when significant maldistribution in the catalyst bed is measured), it can make sense to unload, screen and reload the catalyst.
Although it has been performed successfully, more often it has led to more pressure drop problems.
Best practices:
First, the safety aspects of handling a pyrophoric or self-heating material MUST be addressed. We believe it is critical to use proper detectors (SOX, H2S, etc.) and have proper emergency procedures and properly trained personnel in place before executing the catalyst dumping, screening and reloading process.
It is also important to dry the catalyst before unloading. Dust and small particles stick to the oily catalyst during dumping and screening and are not removed until liquid washing of the catalyst surface. This can lead to fouling of the catalyst bed and reactor internal trays and create excessive pressure drop at restart. H2 stripping to remove liquid between particles before unloading the catalyst is highly recommended.
It is important to split the different catalysts layers by either vacuum unloading guard and grading layers from the top or size screening after dumping in order to be able to reload a properly designed catalyst system.
If the screening is done on-site, we recommend the use of a relatively large screen to ensure broken fragments and small particles are rejected. This will prevent differential pressure issues with the reloaded material.
Providers of onsite screening services are typically limited in available equipment compared to offsite specialized companies. It is important to have adequate equipment to determine the particle size distribution of the screened catalyst. Evaluating the entire length distribution and not just the average length is a critical step for preventing excess pressure drop. It is also advisable to conduct a pressure drop test on the pilot scale to fully evaluate the effectiveness of the screening and predict the corresponding pressure drop for the screened catalyst load. Often some additional fresh guard, grading and main bed catalyst is needed to ensure a complete reactor fill.
Martin Gonzalez (BP)
Dump, screen, and re-load of spent hydrotreating catalyst are usually unattractive for various reasons. To minimize oxygen exposure, catalyst loading companies can usually keep screening equipment under inert atmosphere. Vacuuming catalyst rather than dumping can help to simplify screening by avoiding a mix of many sizes of material. Modern vacuuming equipment can also result in less breakage of catalyst, compared to gravity unloading.
Tim Lewer (Shell)
The practice of unloading, screening, and reloading still-viable hydrotreating or hydrocracking catalysts during turnarounds and without off-site regeneration has been done successfully – with catalyst vendor on-site support or project management. However, there are numerous factors involved in the decision to do so, especially with the advent of stacked bed catalyst schemes with multiple layers of materials either equal in size or too close in size to screen successfully using typical mobile equipment. For single bed hydrotreating reactors, which often contain a majority of same type/size catalysts, the task is more manageable but still a complex undertaking.
Extensive pre-planning is paramount to the success of this type of operation. There are a number of “best practices” that will help to ensure a successful outcome after the decision is made to re-load unregenerated catalysts versus loading fresh catalysts:
1) Research and hire a catalyst handling contractor that possesses the latest catalyst vacuuming/N2 recirculation, dust control, and screening equipment. All equipment used in such an endeavor must have the capability of being effectively purged with nitrogen.
2) Vacuuming support/grading layers from the tops and bottoms of beds and keeping these materials separate from main bed catalysts will allow for quicker screening rates and cause fewer losses due to breakage.
3) Ensure that there are excellent QA/QC measures in place, such as container labeling (where in the bed material is from, pre- and post-screened tare and net weights of all containers, etc.) and strict, written tracking and segregation of unscreened and screened containers.
4) It is imperative to establish clear guidelines prior to the start of the project to ascertain what properties need to be met in order for screened catalysts to be acceptable for reloading. These guidelines should include acceptable levels of fines, average catalyst length, and acceptable levels of cross contamination between different catalyst types and/or sizes. Screen sizes and opening shapes (slotted or square), controlled rates and constant oversight by personnel aware of the established guidelines and desired outcomes of the screening operation are critical to a successful end product.
5) Consult with your catalyst supplier to arrange for lab testing of unloaded materials to ascertain viability of the catalyst and be prepared to perform average length testing on-site (pre and post screening) to ensure catalyst to be reloaded will meet preset guidelines and properties and perform as desired. Catalyst and support losses due to breakage and attrition must be anticipated with assurance that ample make-up quantities of all materials are in ready supply and available as needed. It is also wise to have a “Plan B” prepared in the event that results are less than desired.
6) Pre-plan for sufficient acreage to accommodate a high traffic operation with requirements for staging of empty, unloaded (full) containers, weighing (pre- and post-screening), and staging of materials for reloading. Screening areas should also have weather protection constructed prior to the start of screening operations.
Greg Rosinski (ART)
Spent hydroprocessing catalyst is pyrophoric due to small particulates of iron sulfide scale that are present, so care must be taken to minimize the exposure of the spent catalyst to air. In addition, spent sulfided catalyst has some coke on it and it will slowly oxidize in air. If the spent catalyst is exposed to air, it will slowly heat up, and if iron sulfide is present, it will combust which may ignite the coke or other residual hydrocarbon on the catalyst.
The key to this procedure is to have competent and experienced personnel performing the required tasks. The reactor must be thoroughly swept of hydrocarbons, and a nitrogen purge should be kept on the reactor at all times. During the unloading, the screener and the dump nozzle should be continuously purged. The containers that will hold the catalyst during unloading should be blanketed with nitrogen or have dry ice placed inside until ready for loading. The containers should not be open to the atmosphere. The loading should be done under inert conditions with experienced personnel.
When preparing your procedure, make sure to involve your refinery EH&S group and give careful consideration to all aspects or the process to ensure you take all the precautions necessary.