
Question 11: What constitutes adequate quench reserve when you process cracked feedstocks in hydrotreating units? In hydrocracking units? What if a mixture of both gas and liquid quench is used?
WENDY WILDENBERG (Flint Hills Resources)

We will start out this session on Hydroprocessing with an analogy to begin to provide the answer.
Have you ever watched a child walk a dog? Maybe a big dog?
The dog may be obedient and staying with the child for a while – as long as the dog wants to go the direction that the child is leading. The child has the illusion of being in control of the dog.
But if the dog gets distracted – say by a passing squirrel – the dog may take off and hurt the poor child.
Maintaining control means that the dog walker – needs to be able to hang on to the dog – even if it starts to walk or run away. This takes adequate reserve strength - and an early detection and response when the dog just begins to become distracted or when we begin to feel the pull on the leash.
Same with a hydroprocessing reactor. We need to maintain enough recycle hydrogen to provide sweeping of the oil and cooling of the heat of reaction. We need to provide adequate reserve cooling to handle the size of the reactor duty that we allow. Hydrocrackers are big dogs – and ready to pull away and run. Hydrotreaters will walk away until the temperature reaches Hydrocracking temperatures (>800°F).

At that point – we have the increased duty of a Hydrocracker running away – we have measured accelerating at over 10°F per second increase. This slide shows the acceleration measured on a highly active zeolite cracking bed when the quench was decreased by a faulty temperature indication.

Hydroprocessing Units therefore need reserve cooling and good detection and control to be able to regain control of a reactor and stabilize the temperature in case of higher heat of reaction. One addition of increased heat of reaction is more feed olefins. A unit that has more coker feedstocks, with swings or increases in these olefinic feedstocks will require higher reserve cooling duty to compensate than a virgin hydrotreater that has hardly any change in exotherm from feed changes.

A simple definition of a temperature excursion is that the Heat of Reaction is increasing at a rate faster than the ability to increase cooling to balance the reactor. Unlike our dog story – our reactors increase in size of reaction duty as they increase in temperature. It is harder to regain control once they start to get away.

Several causes of increased heat of reaction for your unit during normal operation need to be considered when determining the amount of reserve cooling or quenching.
-
INCREASE in Cracked Stock (increase in olefin and aromatics)
-
INCREASED TIME in REACTOR (partial loss of feed or reduced feed rates)
-
INCREASE in reactor temperature (planned or unplanned like heater over-firing)

Considerations for Hydrotreaters vs. Hydrocrackers:
Differences between Hydrotreating and hydrocracking (catalyst contains acid function) exist that must be accounted for in determining where to maintain reserve quench and determination of safety interlocks / response.
Hydrotreaters can have temperature excursions, but there are a few other factors to consider that allow reserve cooling to be applied in other areas as well or even instead of the reserve quench. Until the high temperature is reached, hydrotreaters will “walk away”. Therefore, reducing firing / tripping the heater fuel gas and stopping feed may stop the growing heat wave in time to prevent temperatures from exceeding design limits.

Hydrocrackers have higher heat of reaction from saturation reactions as the cracking function creates olefins that are then saturated. There are plenty of reactants available. Reactor catalyst beds with cracking catalyst must have the ability to apply reserve cooling directly to the bed as well as the ability to cool from upper beds. We have witnessed from a sudden loss of quench, hydrocracking beds rising from normal temperatures to > 1500°F in that bed.

Reserve quench hydrogen provides direct cooling to the individual bed for the fastest response and is required for all hydrocracking beds. Best practice for quench hydrogen is to measure the hydraulic capability and then hold 50% of that capability in reserve. Use caution if setting limits for normal quench on % opening of a control valve. If the control valve has equal percentage trim, 70% open may be near 100% of hydraulic capability. Also ensure that the control system is designed to use the full capacity of the control valve, and not limit the valve opening based on the flow meter span.

Other cooling knobs that may be applied to reduce the temperature of the reactor include Reserve compressor capacity, feed oil bypass, liquid quench (includes recirculation of product to feed) and reductions in charge heater firing.
Cold oil may be sent to the heater and/or reactor inlet by bypassing the feed/effluent heat exchange. To ensure adequate reserve is maintained, measure hydraulic capability with valve fully open to determine range of cooling available. Beware of the design of low exchanger train pressure drop with a simple control valve bypass. As the bypass control valve is opened, the driving force for flow through the bypass decreases leading to lower hydraulic capability for providing cooling.

The higher this ratio, the more the unit is capable of regaining control with heat of reaction increases.
Reserve duty is defined as capacity not used for normal operation, but capable for use. Best practice is to set normal operational limits based on % of capacity, not on % opening of control valves.
An important point I need to share in case of recycle compressor failure, no reserve capacity for quench is available. Use Risk assessment in your units to determine procedural and safety interlock responses necessary.
Year
2018
Process
Question 12: Discuss impacts of hydrotreating operations required to meet Tier III regulations. Highlight the benefits and concerns of pretreat versus post treat operations including; impacts on cycle length, FCC yields, octane from post treating options, and gasoline blending.
JEFF CATON (Axens)
Regulatory specifications for the gasoline and diesel pool, which are constantly evolving, have been in the forefront of refiners’ challenges in the last 15 plus years. In particular, the gasoline sulfur regulations have been a main driver for the remodeling of many refineries’ configurations. Specifically, in the US, most refiners have already addressed, or are continuing to address, the challenges to meet the US Tier 3 ultra-low sulfur gasoline (ULSG) <10 ppmw sulfur specification, which became effective on January 1, 2017. Most US refineries now have FCC pretreatment (CFHT) and/or FCC post-treatment unit(s) to ensure compliance with Tier 3 requirements (or Tier 2 requirements for refineries with waivers).
When the US Tier 2 gasoline regulations were initially proposed in the late 1990s and subsequently implemented in the 2000s, many refiners were convinced that CFHT would be the solution of choice. In addition to producing LSG/ULSG and improving FCC performance, additional benefits of volume swell through aromatics saturation and a reduction of FCC SOx and NOx emissions could be realized. A major benefit to producing LSG/ULSG with CFHT alone, with no post-treatment, is the retention of olefins in the FCC gasoline, thus benefiting the overall refinery pool octane balance.
Within the context of producing ULSG, the primary role of CFHT is that of desulfurization and the impact on the FCC gasoline produced. If one were to target a ULSG pool sulfur level of 10 ppm, the CFHT must reduce the FCC feed sulfur to about 200-300 ppm, considering a typical ratio of between 20:1 and 30:1 of the hydrotreated FCC feed sulfur to the FCC gasoline sulfur. When looking at the sulfur in FCC gasoline, the gasoline cut point and the distillation tail on the produced gasoline product is of high importance. In the US market, gasoline has been traditionally over-cut relative to the standard 430°F (220°C) cut-point and often extended to 450-480°F (230-250°C), thereby including not only benzothiophene but also some methyl-benzothiophenes in the gasoline (Refer to Figure 1 - FCC Gasoline Sulfur Profile). By over-cutting, these more difficult sulfur compounds, which will remain at a significant level following CFHT, will subsequently find their way into the FCC gasoline and make it difficult to produce ULSG. Undercutting the gasoline to less than 430°F will significantly help control the sulfur level when producing ULSG.
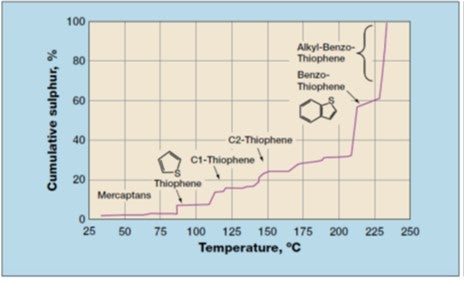
Considering the dependence of FCC gasoline on both CFHT performance and the need for precise fractionation of the FCC gasoline product, meeting ULSG targets through CFHT alone is possible, but challenging. There is little room for error or deterioration in CFHT performance over the course of a production run or cycle. Further, there can be limited flexibility in crude slate, thus reducing the refiner’s ability to process opportunistic crudes. These deterrents along with the relatively high capital cost for CFHT coupled with low refinery margins have resulted in the wide application of FCC post-treatment to reduce FCC gasoline sulfur. In fact, many refiners with CFHT have found it necessary and/or economically advantageous to also have a post-treatment unit.
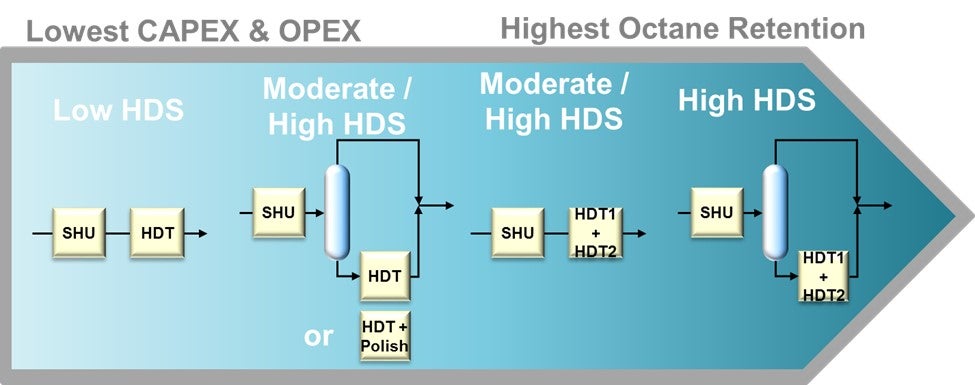
Most refiners have already invested in an FCC post-treatment unit. For a refinery with relatively high sulfur FCC gasoline, the post-treatment severity will be higher in order to produce ULSG (Refer to Figure 2 - Prime-G+TM Designs at 10ppm Product Sulfur). This higher severity can lead to an unwanted high-octane loss through the undesirable saturation of olefins in the post-treatment unit. However, depending on process technology and process flow scheme selected for the post-treatment unit, the concern over octane loss can be managed. Axens Prime-G+ process technology has been applied extensively with varying flow schemes (with corresponding varying capital cost investment) to produce USLG with an octane loss that is within the constraints of the refiner’s pool octane balance (Refer to Figure 3 - Prime-G+™ Tailored Scheme). In short, a simplified process flow scheme with a single reactor combining selective hydrogenation (SHU) and hydrodesulfurization (HDS) can be utilized when FCC gasoline sulfur is relatively low while yielding a very low octane loss. Conversely, a more complex process flow scheme with a SHU reactor, splitter, and HDS reactor can be utilized to treat a higher sulfur FCC gasoline while still minimizing octane loss to within the constraints of the refiner’s pool octane balance.
Figure 2 - Prime-G+TM Designs at 10ppm Product Sulfur
Figure 3 - Prime-G+™ Tailored Schemes
Where a refiner is producing ULSG through CFHT alone, the CFHT’s operating severity will be heavily dictated by the CFHT feed sulfur. In this scenario, if the refiner were to purchase a more opportunistic heavier and higher sulfur crude, there would be a resultant CFHT severity increase and a negative impact to the CFHT’s cycle length. Depending on the CFHT’s original design constraints, there may even be limits on what crude slate can be ran while still allowing production of ULSG. Similarly, with post-treatment alone, there would be an increase in post-treatment feed sulfur thus resulting in an increased severity requirement in the post-treatment unit. This would directionally have a negative impact on the post-treatment unit’s cycle length and would also result in a higher level of olefin saturation and corresponding octane loss. Having both CFHT and post-treatment, there is more potential to balance the overall higher severity requirement between both units thus having a lesser impact on cycle lengths and octane loss. Further, with both CFHT and post-treatment, through feed management and altered operating conditions, it may be possible to continue operations and production of ULSG while either the CFHT or post-treatment unit is off-line. Unfortunately, having both a CFHT and post-treatment unit comes at a high capital investment, and this can be a prohibitive barrier.
WENDY WILDENBERG (Flint Hills Resources)

I can share two different approaches of use of hydroprocessing to meet Tier III regulations within the Flint Hills refineries.
A southern refinery has both an FCC Feed hydrotreater as well as an FCC Gasoline hydrotreater. Upgrades to the gasoline hydrotreater have been made in the last several years to achieve higher capacities and severities to lower sulfur levels in gasoline. The FCC Feed Gasoil hydrotreater has a lower severity, may hydrotreat 40 to 90% of the FCC Feedstock – and may even be bypassed if gasoline hydrotreating can be increased for lower opportunity cost maintenance as needed. Attention is necessary to balance the severity of the pre-treatment and post-treatment as Jeff mentioned to balance yield, severity, and runlength of the FCC Pretreater with olefin/octane loss of the post-treater. Feedstocks can be chosen to be processed in the Gasoil hydrotreater to maximize yield of the GOHT/FCC combined while allowing the easier feedstocks to feed the FCC directly.
Prior to the Tier II regulations, our northern refinery was already hydroprocessing 100% of the FCC feed to meet environmental emissions targets as well as for the increased LV yield. Severity increases were necessary to meet the lower sulfurs of Tier II and Tier III since the FCC gasoline is not further processed before gasoline blending. Improvements in catalyst activity were beneficial to lowering start-of-run temperature as well which is very helpful to increase runlength. Established guidelines and attentive monitoring for key parameters such as feed line-ups, hydrogen recycle rate, hydrogen purity, and temperature adjustments allow this refinery to process a large concentration of opportunity feedstocks and achieve longer runlengths (3-4 years). In this refinery, not only is the FCC Gasoline octane maintained without further processing, but the slurry oil sulfur has been < 0.5 wt% for the last several years.
GEORGE HOEKSTRA (Hoeskstra Trading LLC)
Hoekstra Trading sponsored a three-year program of pilot plant testing, catalyst testing, field testing, and market analysis, all aimed at answering the question, “How much octane will be destroyed when post treating severity is increased to make Tier III gasoline?”
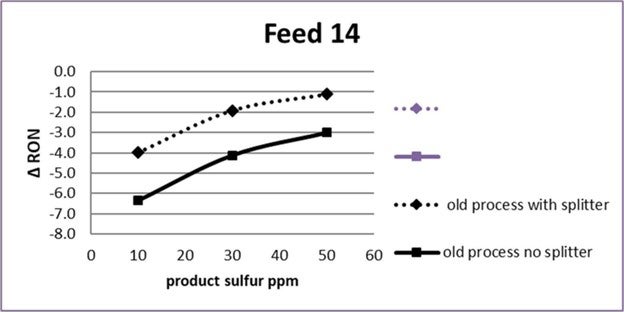
The answer from our pilot plant testing is displayed in what we call the post-treater “performance curve” which is a chart of octane versus product sulfur:
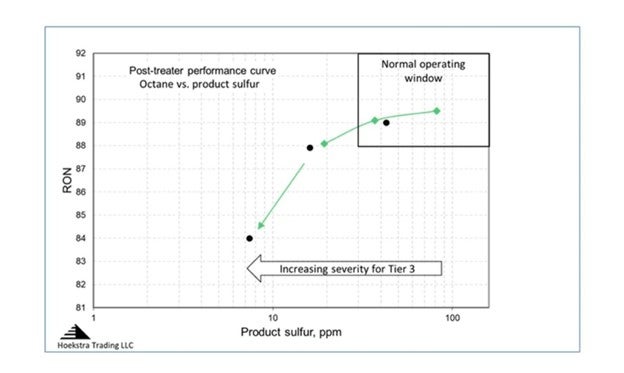
In this chart, post treater reaction severity is being increased as you move from right to left. As severity increases, gasoline sulfur decreases, and so does octane. This feed is from a US refinery, it is a full-range FCC gasoline with 443 ppm sulfur that has been pretreated in a diene saturator. The green and black data points are for two different catalysts. The normal operating window for post-treaters has historically been in the upper right region of the chart where the curve is flat, meaning little octane is lost as you increase severity to decrease product sulfur; but now, we are moving left into a new operating region for Tier III gasoline where the rate of octane loss accelerates. The acceleration is much higher than the industry expectation which says that octane will go down by 0.5 RON when severity is increased to make Tier III gasoline. Our pilot plant data shows it is much higher than that, which was an important new finding in 2015.
The next step was to see whether this important finding holds true in commercial post-treaters in North America. Our client group has run detailed chemical analyses on post treater feed and product samples from field tests on eleven commercial FCC gasoline desulfurizers. The results confirm our pilot plant findings — commercial post-treaters also lose 5 octanes when severity is increased to make Tier III sulfur gasoline.
Not all refineries will lose 5 octanes, only those that are not well-equipped to make Tier III gasoline reliably and profitably. The octane loss in each case depends strongly on the configuration of the FCC process train, the design of the post-treater, and the composition of the raw FCC gasoline. We have seen octane loss over 10 on some commercial units. It is highest for units that have no FCC pretreater and that run heavy, high sulfur feeds.
This chart shows how the performance curve will change for one refiner who decided to add a splitter to his post-treater for Tier III gasoline.
The dashed curve shows, at 20 ppm product sulfur, the octane loss will be 3 with the new splitter, compared to 5 (solid curve at 20 ppm) today.
These curves can be shifted up by cutting heavy tail out of the post treater feed. This chart shows the performance curve in the form of a spreadsheet model that is being used today by refiners and process licensors to estimate and optimize post treater performance for different feeds, process designs, and operating strategies. The dashed curve shows that, with a splitter and endpoint cutting, octane loss can be reduced to 2 RON (dashed curve at 20 ppm sulfur).
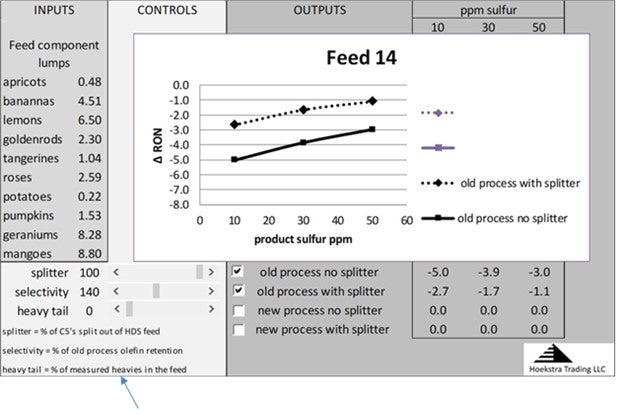
The controls at the bottom left of the worksheet permit varying the splitter efficiency, the heavy tail percentage, and the process selectivity to see the effects on the performance curves.
It is possible, with new catalysts and processes, to improve sulfur/octane selectivity by as much as 40% from historical levels. The purple curves show this effect:
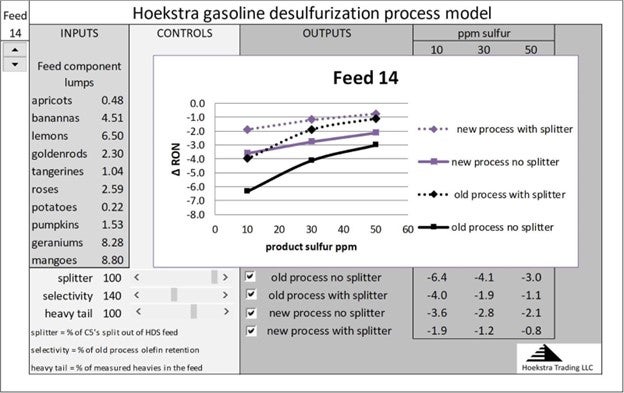
This full range of process improvement is available to refiners today through improved process design, catalyst selection, and feedstock management.
These curves are calculated accurately for any unit by entering the analytical data for that unit’s feed into the spreadsheet model. The model uses reaction rates that have been measured in pilot plant and commercial field tests on real feeds over a wide range of conditions.
The spreadsheet model and all the pilot plant and commercial test data are available to anyone for a small fraction of the cost of this work, which has been done by our client group over the last 3 years.
Year
2018
Process
Question 13: What factors do you consider when co-processing jet fuel in a distillate hydrotreater versus processing the jet separately (including feedstock and unit consideration)?
ROBERT STEINBERG (Motiva Enterprises)
There are several considerations when deciding if jet fuel and diesel should be co-processed or hydrotreated separately. The most important consideration is if the jet fuel will be blended into the diesel product or if separate products are desired. The decision may depend on if you are looking at constructing new facilities or making the best use of existing equipment.
If a refiner needs to build a new unit to increase distillate hydrotreating, building a jet fuel unit and using existing units for diesel will normally be less expensive than building a new diesel hydrotreater. If more capacity is required than there is jet fuel for, some straight run diesel can be blended into the jet fuel hydrotreater. However, if the existing unit is too low a pressure to handle the diesel effectively it may be preferred to build a new unit for the most difficult to treat streams such as light cycle oil or light coker gasoil instead.
If a new refinery was being built and new jet fuel and distillate hydrotreaters are needed it will normally be simpler and cheaper to build a single unit and co-process the jet fuel with the diesel. Exceptions would be if the capacity would be too large for a single unit or separate jet fuel and diesel products were desired.
For a refiner looking to optimize existing facilities it is assumed that a separate jet fuel product is not required. If it was required, the feeds would need to be processed separately unless there was a fractionator on the back end to make jet fuel and diesel cuts. Having such a fractionator is probably unusual and would require a lot of energy to vaporize the jet fuel to remove it from the diesel. For a refiner with only a single hydrotreater this becomes a question of batch processing diesel and jet fuel or blending them and processing together. If the refiner has multiple hydrotreaters it will generally make sense to send the easiest to treat streams, such as jet fuel, to the lowest pressure or mildest hydrotreater. Or blend just enough of the easiest streams into the more severe unit to let it achieve its desired run length.
Alternately batch processing jet fuel and diesel in the same unit instead of co-processing would generally not be recommended unless there were special circumstances. This would be more complicated as it would require frequent switching of feeds and changing operating conditions; during such changes lower charge rates may be needed to reduce the risk of making off-spec products or having to pull more naphtha out of the Stripper than otherwise required to maintain product flash point.
Diesel requires more severe reactor conditions than jet fuel to make the same product sulfur. Mixing jet fuel into the distillate hydrotreater lowers the average boiling point of the feed as well as the feed sulfur and nitrogen content. This means lower reactor temperatures and less chemical hydrogen consumption, less treat gas is needed to maintain the desired ratio of hydrogen availability to consumption, smaller exotherms and less quench gas. The lower start-of-run temperature can extend the catalyst run length.
Pressure drop for jet fuel and diesel in the same hydrotreater would be similar if the same amount of hydrogen circulation was used – more of the jet fuel would vaporize and increase velocity but the diesel has more mass at the same barrel per day charge rate and is more viscous. However, less treat gas is needed for jet fuel due to the lower chemical hydrogen consumption and exotherms are generally low enough with jet fuel to not need any quench. This effectively means less hydrogen circulation with jet fuel and lower pressure drop in the reactor, exchangers and furnace. Blending jet fuel into the diesel hydrotreater will reduce pressure drop, if the run length is limited by pressure drop this can extend the catalyst life.
Another consideration can be product blending and diesel cetane. If there are multiple units, or jet fuel and diesel are batch processed, some of the products may not meet sulfur, cloud point, cetane or other product specs. In some cases, the products from different units can be blended to make an on-spec ULSD. This makes operations more complicated, adjusting the amount of jet fuel that is blended into each unit can help keep all products on-spec and reduce the risk of having to deal with an off-spec product tank. However, jet fuel has a lower boiling point than diesel and thus a lower cetane number. If a refiner makes both a higher and a lower cetane product it may be necessary to minimize the jet fuel in the high cetane product, this is like making a separate jet fuel product in that it can require separate processing.
Motiva has a relatively mild hydrotreater than was revamped for ULSD 15 years ago. Initially it charged mostly straight run diesel to make ULSD. Later, the refinery was expanded, a new diesel hydrotreater was built and the additional jet fuel was charged to this unit. The unit now charges only jet fuel and operates in ultra-low sulfur kerosene (ULSK) mode, the product has less than 5 ppm S and can be blended into either jet fuel or diesel as desired. The unit has minimal catalyst aging. It illustrates that jet fuel can be the easiest product in the refinery to hydrotreat giving very long catalyst life and that adding jet fuel to a diesel hydrotreater results in milder reactor conditions and longer catalyst life.
Motiva normally produces more kerosene in our crude units than we have capacity for in our jet fuel hydrotreaters. The surplus kerosene is mixed into our diesel hydrotreaters. This is the easiest feed the diesel hydrotreaters process, with more kerosene reactor temperatures can be lowered while maintaining product sulfur. While this reduces the required reactor severity there are some drawbacks:
-
The lighter feed reduces the delta API gravity between feed and product meaning volume swell is reduced.
-
The lighter feed reduces the density of the feed going through the charge pump and the discharge pressure that the pump can produce. One of the diesels hydrotreaters sometimes needs to lower operating pressure to maintain charge rate.
Motiva normally hydrotreats as much kerosene as there is capacity for in our jet fuel hydrotreaters and blends the remaining kerosene into diesel hydrotreaters. One of the jet fuels hydrotreaters swings between sending its product to jet fuel and diesel but operates to be on-spec for both products. We do not have any units where we switch feeds or operating conditions to sometimes make jet fuel and sometimes diesel.
JOHN KULACH (UOP)
Considerations for co-processing jet and distillate in the same unit are the feed rates and feed quality, which go into the selection of operating pressure, space velocity, and catalyst. Distillate hydrotreating for ULSD typically requires higher hydrogen partial pressure, lower LHSV and more active catalysts compared to treating jet, because of the need to convert stable sulfur compounds such as benzothiophenes and dibenzothiophenes. If the distillate feed includes coker gas oils, LCO or other difficult to process streams such as extracts and condensates, the design would call for a more severe operation with higher reactor temperatures. Inorganic contaminants such as silica and arsenic can be removed from the feed upstream of the active desulfurization catalysts by using filters, particulate traps, and demetalization catalysts in the top of the reactor.
Jet hydrotreating requires enough catalyst and hydrogen partial pressure for mercaptan sulfur and TAN removal. This is usually a low severity operation compared to distillate hydrotreating as jet fuel can be very color sensitive exasperated by high reactor temperature operation. On the other hand, jet fuel hydrotreating can require more severe operation if the feed contains high aromatics or naphtenes which will require some saturation to meet composition and combustion specifications such as aromatic content and smoke point. In some cases, this might require a noble metal catalyst or a second stage operation.
While a typical distillate hydrotreater fractionation section would consist of a stripper column to remove light ends and meet flash point specification, a hydrotreater co-processing distillate with jet feed would require a more complex fractionation section. The fractionation configuration would depend on the relative rates of distillate and jet as well as the need to meet jet fuel volatility and fluidity specifications such as distillation and freeze point.
Whether a co-processing unit is more economical than individual DHT and KHT units will depend on the relative feed rates, feed quality and product specifications. Co-processing might be favored if the DHT feed is relatively easy (such as a SR Diesel) and if the jet feed is more difficult to treat. The key take-away is that jet fuel processing is very dependent on the feed quality and required specifications.
RICHARD TODD (Norton Engineering)
Co-processing of Jet Fuel in a distillate unit may result in poorer unit performance than expected due to higher vaporization of the jet fuel components, which in turn causes lower H2 partial pressure. This varies based on unit conditions, partial pressure of H2, LHSV, etc. The heavier distillate desulfurization is normally controlling, so reactor temperatures are set by the distillate requirements. Cracked stocks in the jet fuel range may have higher olefins increasing H2 consumption, and again in turn, decreasing H2 partial pressure, causing an increase in deactivation.
Year
2018
Process
Question 14: In your experience, what operational factors contribute most to utility costs in hydrotreating units?
ROBERT STEINBERG (Motiva Enterprises)
The biggest contributor to utility cost per barrel in a hydrotreater is probably the unit operating pressure. Higher pressure units require more energy to pump up the charge, hydrogen, amine and wash water.
The utility cost that may get the most attention and that operations has control over is heater firing. Use of a hot separator and bringing in hot feeds can substantially reduce heater firing. However, fired duty per barrel of charge is generally much lower for a hydrotreater than for many other refinery units like crude units, cokers and reformers.
Some people like to keep H2 to oil ratios to the minimum required for the catalyst even if they can circulate more. While this will save some energy, it may not be optimal for the catalyst. Using more than the minimum required recycle gas will increase H2 partial pressure and may reduce catalyst aging rates, it can also improve distribution within the catalyst bed. If catalyst life is extended, yields are improved, or product properties are improved the benefits can be much larger than the energy savings from the minimizing H2 to oil ratio. In addition to the catalyst benefits, higher recycle gas rates will increase velocities in exchangers and furnaces and may reduce fouling.
Some hydrotreaters maintain the same charge rate all the time using product recycle to maintain reactor feed if less fresh feed is available. It is necessary to maintain reactor charge rates high enough to be within the operating range of the distributor tray. If there are large amounts of cracked feeds, recycle can be needed to control exotherms. Maintaining the same charge rate can simplify operations, reduce exotherms and improve distribution within the reactor without impacting required reactor temperature or catalyst aging rate. However, it can also substantially increase energy consumption. For a short-term reduction in charge rate, such as for a few days, the simplicity of maintaining operating conditions may justify the recycle. For longer time periods, such as several weeks, the impact should be evaluated more carefully to see if more recycle than needed to maintain minimum charge rates is justified.
Using more than the minimum recycle gas rate can often extend catalyst life at the cost of a marginal increase in energy consumption. Using excess product recycle will often have little benefit and substantially increase energy consumption.
If there are advantages to using more than the minimum recycle gas or product recycle, the benefits such as better catalyst performance and simplicity should be compared to the extra energy cost to maximize profitability.
Year
2018
Process
Question 15: As hydrotreating catalyst development continues with the emphasis on activity and saturation, how has this changed optimization strategies for the entire hydrocracker?
NEIL HOWARD (Chevron Lummus Global)
Advances in hydrotreating catalyst technology continue to produce steadily increasing performance for hydrocracking pretreat service. These improvements result in increased HDN and aromatics saturation performance which are key to optimizing overall hydrocracker performance.
There are many ways this improved performance can be utilized:
-
Processing lower cost, more difficult to treat feedstocks
-
Targeting deeper HDN to install lower activity, more distillate-selective hydrocracking catalysts
-
Taking advantage of lower start-of-run temperatures to target longer cycle length
Processing more difficult feedstocks
Improvements in pretreat catalyst activity has steadily continued over the last several years, which has made it possible to send more profitable feedstocks to the hydrocracker. This can be through more refractory feed selection, or simply increasing the boiling range of the hydrocracker feedstock by cutting deeper in the vacuum distillation unit.
Below is an example which shows an increasingly heavier feedstock with each progressing catalyst cycle made possible by more active pretreat, as well as a more nitrogen tolerant cracking catalyst system. In this case, the refiner’s crude source has been reasonably constant, but advances in the pretreat and the cracking catalyst systems have allowed them to progressively process heavier feeds with each subsequent cycle by going up in feed endpoint.
Table 1: Feed Basis Over Subsequent Cycles


Increased options for cracking catalyst selection
More active pretreat systems also allow for more flexibility in cracking catalyst selection. For example, by going to a more active pretreat system, it is possible to trade off this activity gain with the use of a more middle distillate selective catalyst in the cracking system. Below is an example of a typical progression of this nature. In this case, the refiner is targeting maximum middle distillate within the constraints of a minimum 30-month cycle length. By installing a more active pretreat system, the refiner can make use of a more middle distillate selective cracking catalyst in one or more beds of the hydrocracking reactor. The impact of going to a more distillate selective catalyst in the last bed of such a system is significant, as the selectivity improvement in this highest conversion regime of the hydrocracking reactor is most pronounced.
Figure 2: Increasing the Pretreat Activity with Newer Generation Hydrotreating Catalyst
The increased activity of the pretreat system changes shown above enable the replacement of one bed of hydrocracking catalyst in the bottom bed of the hydrocracking reactor with a more middle-distillate cracking catalyst as shown below:
Figure 3: Increased Pretreat Activity Allows More Distillate Selective Cracking Catalyst
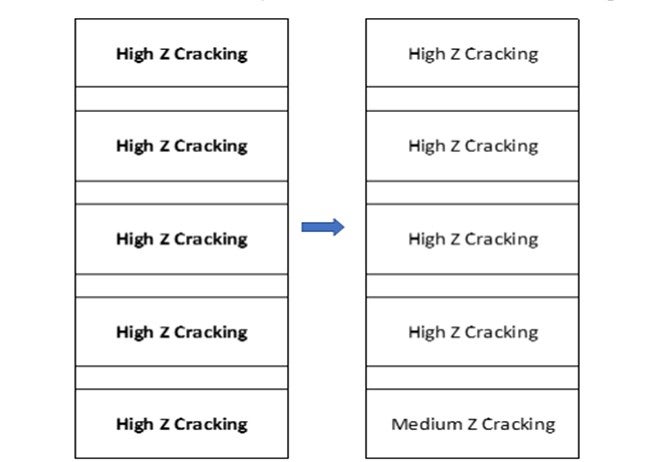
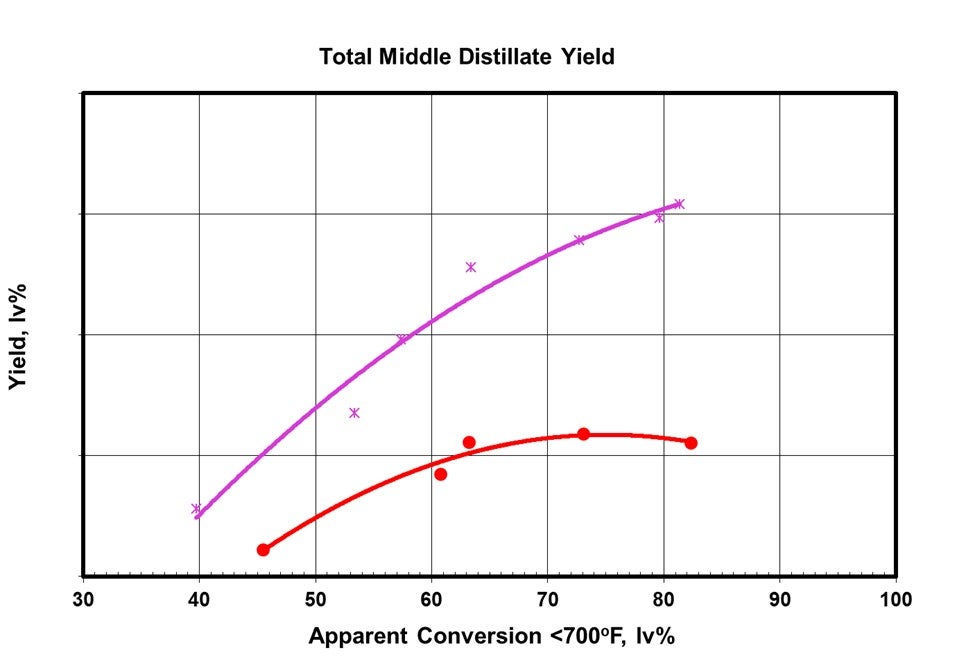
This one bed of more mid-distillate selective catalyst buys a significant improvement in middle distillate yield by virtue of its placement in the highest conversion environment in the reactor. It can be seen from the following comparison that the selectivity advantage of going to a somewhat lower activity, more mid-distillate selective catalyst, in the bottom bed results in a large boost in the marginal increase in middle distillate in this range of 60-70 lv% conversion.
Figure 4: Incremental Selectivity to Middle Distillate
Self-Supported very-high activity catalyst options
Many catalyst vendors now offer very-high activity hydrotreating catalysts that are more often finding use in hydrocracking pretreat systems. These catalysts have much stronger aromatics saturation, HDN and HDS activities than conventional alumina-based catalysts. As discussed above, this activity improvement can be used to process more difficult feedstocks or take advantage of more catalyst flexibility because of the activity improvement in the pretreat section of the hydrocracker. Or some refiners find an economic payout in the form of longer cycle length afforded by such catalysts.
Below is an example of this concept in which a very-high activity self-supported catalyst is used to enhance the activity of the pretreat system to extend cycle length of the entire hydrocracker. The key to this sort of optimization is to appropriately size and place the self-supported catalyst to extract the maximum benefit. Because of the very high aromatics saturation activity, placement of such catalysts too high up in the pretreat system will result in excessive coking, which can prematurely deactivate the catalyst, and in some cases, make the catalyst difficult to unload due to hard coke ball formation. The sizing and placement will depend upon the feedstock and processing objectives.
To extend our example above, here is a case in which a portion of the pretreat system is replaced with a very-high activity self-supported hydrotreating catalyst.
Figure 5: Very-high Activity Self-supported Pretreat Catalyst System
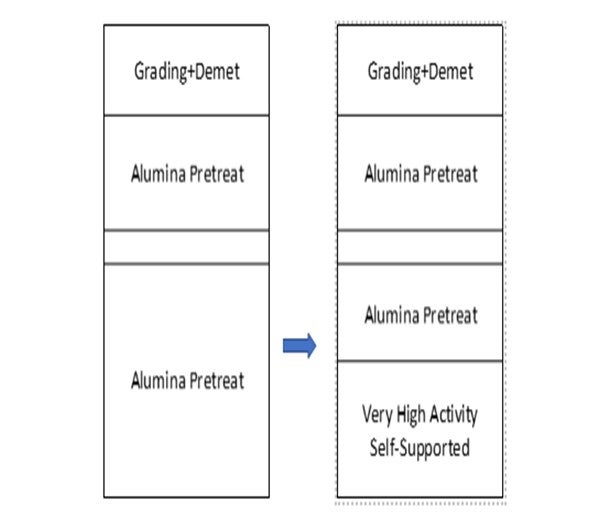
In this case, the resulting activity improvement allows a cycle length increase from 30 months to 36 months on the overall hydrocracker.
Optimization of Pretreat vs. Cracking LHSV
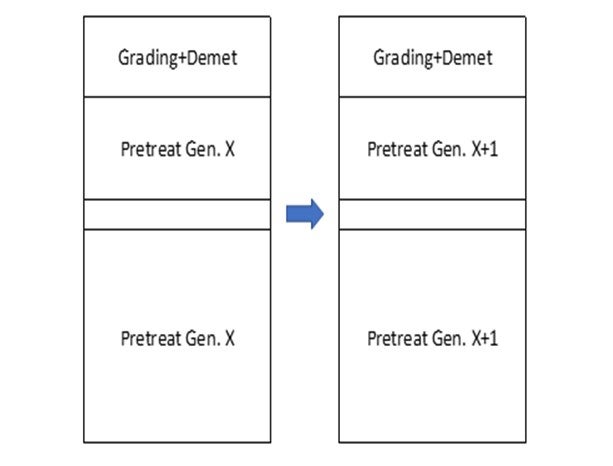
In addition to the examples above, many refiners will achieve more profitable operation by shifting the ratio of pretreat to cracking catalyst volumes with changes in process conditions or objectives. For example, it isn’t uncommon to trade out pretreat volume for cracking volume in cases where cracking activity is limiting. This type of adjustment is made possible by higher activity pretreat catalysts maintaining performance at higher LHSV. This can allow a refiner to maintain a more distillate selective catalyst system than would otherwise be possible with limitations in cracking catalyst volume due to capacity increases, or feedstock changes, over the life of the hydrocracking unit.
There are several considerations in implementing this type of adjustment. Typically, small layers of cracking catalyst can be accommodated at the bottom of the last pretreat bed. However, as this cracking layer is increased it is necessary to be aware of temperature control issues in the bed. To get around this concern it may be feasible to place the cracking layer at the top of the pretreat bed so that the temperature control at the bed inlet directly controls the temperature rise across the cracking catalyst portion of the bed. Alternatively, one can use a co-extrudate catalyst system with combined hydrotreating/hydrocracking functions to uniformly distribute the temperature rise across the entire bed.
The converse of this shifting of pretreat volume to cracking volume is also common for units in which increased capacity, or more difficult feed, necessitates more hydrotreating activity. In these cases, similar considerations come into play with respect to temperature control on a partial bed of pretreat and cracking catalyst. Placing a layer of pretreat atop the first cracking catalyst bed is most common; however, for very deep catalyst beds, a co-extrudate system will provide more stable temperature control.
JEFF CATON (Axens)
In addition to the points already discussed, a higher relative volume activity (RVA) pretreat catalyst can allow for a reduction in pretreat catalyst volume thus freeing up reactor volume for additional top bed grading and/or guard catalyst. For units that have historically had end-of-cycle either dictated by dP or contaminants, this additional grading and/or guard volume can allow for increased cycle length and better monetization of the pretreat and hydrocracking catalyst systems.
There have also been advancements in the area of increased nitrogen tolerance of hydrocracking catalysts (Refer to Figure 1 - Nitrogen Tolerance of New Generation Hydrocracking Catalyst). If there is enough temperature to drive conversion in the hydrocracking system, the temperature of the pretreat system does not have to be increased to maintain a very low nitrogen slip over the entire cycle. By allowing nitrogen slip to drift up towards the end of the cycle, this can provide benefits in terms of maximizing cycle length and/or by optimizing the pretreat system temperature profile in a maximum aromatics saturation mode.
Figure 1 - Nitrogen Tolerance of New Generation Hydrocracking Catalyst
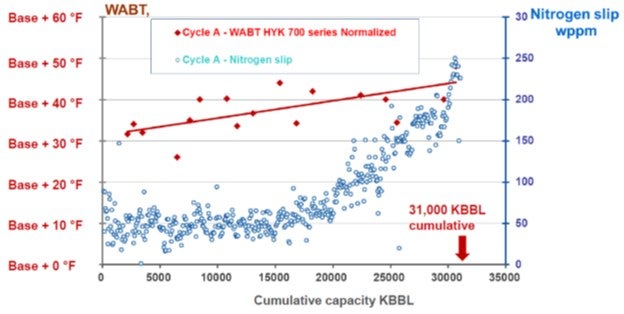
Alternatively, with this increased nitrogen tolerance, the pretreat catalyst volume could potentially be decreased as HDN activity requirement is reduced. Coupling this with current generation higher relative volume activity (RVA) pretreat catalysts, there can be an even more substantial reduction in required pretreat catalyst volume. As discussed previously, this decrease in pretreat catalyst volume can allow for an increased volume of grading, guard, and hydrocracking catalysts.
WENDY WILDENBERG (Flint Hills Resources)
A choice to install catalyst with higher aromatic saturation has allowed the shutdown of a small Jet Aromatic Saturation unit. The Hydrocracker is now able to achieve in the same unit what both units together were accomplishing.
Year
2018
Process
Question 16: How are you performing in-situ sulfiding for hydroprocessing catalysts protecting your waste water units from water soluble organic chemicals and their fuel gas system from non-decomposed mercaptans?
RANDY ALEXANDER and JIM ROBINSON (Reactor Resources LLC)
DMDS decomposes over a CoMo or NiMo catalysts at reactor temperatures in the range of 400-460F, forming H2S and CH4. The exact temperature needed to achieve complete decomposition is dependent on the pressure, the catalyst type, and the space velocity in the reactor. For example, full decomposition is achieved above 430F with a pressure of 300 psi and a space velocity of 1.0 hr-1. At a lower pressure and a higher space velocity, the full decomposition temperature for DMDS will approach 445F.
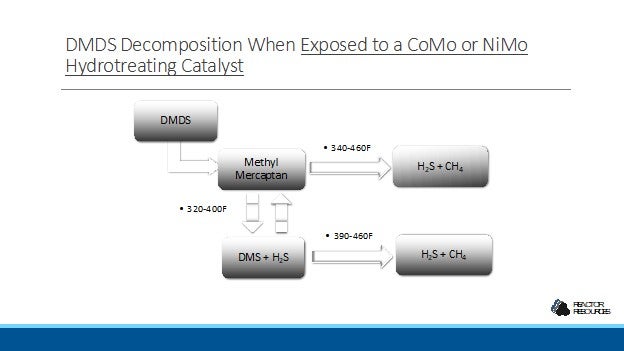
.
There are several approaches that can be utilized in order to avoid issues with non-decomposed mercaptans in wastewater and fuel gas systems.
The easiest solution to this issue is a carefully controlled sulfiding process which begins with updated procedures that include modern control technologies and real-time measurements to manage the process.
Essential control elements include:
-
Real-time H2S measurement.
-
Transparent and accurate real-time sulfiding agent measurement.
-
A reliable means to control the injection rate.
Secondary elements that may also be of benefit:
-
Real-time Hydrogen Purity measurement
-
Total Sulfur measurement
-
Properly deployed, these elements will provide the sulfiding team with enough information to control the in-situ sulfiding activities during start up and avoid the release of non-decomposed (non-H2S) mercaptans to the fuel gas or flare header. This goal can be achieved with any of the sulfiding chemicals normally used (DMDS or TBPS).
It is imperative that the sulfiding team resist the temptation to inject DMDS at a high rate early low temperature stage of sulfiding. Over injection at low temperatures can lead to excessive recycle gas density and low hydrogen purity, driving the need to purge sour gas and add make up hydrogen in order to avoid a compressor trip or catalyst damage due resulting from low hydrogen partial pressure. The sulfiding team should consider moving bed temperatures up to 450F as soon as is practical in order to facilitate full decomposition of mercaptans in the recycle gas.
Purging of sour gas should be avoided if reactor beds are below 450F. Instead, the pressure of the recycle stream should be increased, if possible, in order to facilitate decomposition. if an increase in system pressure is not possible, DMDS injection should be paused while temperatures are increased.
Of secondary concern is the H2S concentration in the recycle gas stream after breakthrough. There is no technical reason to allow H2S concentrations to rise above 10,000 ppm. In fact, any H2S in excess of 1000 ppm is not necessary for good catalyst activation. Levels above 1000 are artifacts of a “safety net” approach developed when older, less timely and less accurate methods of H2S measurement were utilized (e.g., Draeger tubes).
In the absence of the above essential elements, there are other sulfiding chemicals, such as polysulfides or polysulfide blends, that do not produce mercaptans during decomposition. These poly-sulfides, such as di-tert-butyl polysulfide (TBPS), have both positive and negative attributes, but can be used alone or in conjunction with dimethyl disulfide to effectively mitigate mercaptans in fuel gas or flare systems. Full decomposition of TBPS will occur at 325F to form H2S and isobutane. However, due to the lower sulfur density of TBPS (54% versus 68% for DMDS), 25% more chemical will be required to completely sulfide the catalyst bed. Furthermore, due to supply interruptions and erratic market demands, poly-sulfides are typically more expensive and require a longer lead time to be assured of availability.
The last option for avoiding mercaptans downstream of the process, ex-situ catalyst sulfiding, is also the most expensive option. The ex-situ sulfided catalyst is loaded in the active form. Therefore, no sulfur spiking agents need to be injected into the process and the possibility of mercaptans generation during start up is virtually eliminated.
In closing, there are no technical reasons why a modern refinery using current sulfiding techniques should experience issues with mercaptans in the fuel gas or flare header unless there is a unit upset. Over-injection of the sulfiding agent below 450F will exacerbate the problem. Good planning prior in advance of start-up and close attention to detail during injection will typically lead to a successful result.
ROBERT STEINBERG (Motiva Enterprises)
During in-situ catalyst sulfiding di-methyl disulfide (DMDS) or other chemicals like tertiary butyl polysulfide (TBPS) are used to generate H2S to activate the catalyst. These chemicals break down to form H2S and light hydrocarbons. However, the breakdown is not complete. DMDS will decompose to methyl mercaptan and some dimethyl sulfide as well as methane and H2S. TBPS will decompose to tertiary butyl mercaptan as well as butane and H2S.
A small amount of the sulfur in sulfiding agent stays as mercaptans when it leaves the reactor, particularly at initial sulfiding temperatures. While much of these mercaptans are recycled back to the reactor some may leave the unit in:
-
High pressure recycles gas vent.
-
Low Pressure Separator vent
-
Stripper vent
-
Charge Surge Drum vent
-
Sour water from Cold Separator
-
Sour water from Stripper Reflux Drum
-
Stripper bottoms sent to tankage and tank vents.
Gas vents to fuel gas are normally scrubbed with amine before being used in furnaces. Amine will remove H2S but not mercaptans. These mercaptans contribute to total sulfur in fuel gas. H2S analyzers on fuel gas will not detect these mercaptans but SO2 analyzers on flue gas will. Regardless of whether the sulfur is detected it will be present in heater flue gas.
If the mercaptans are water soluble they can leave the hydrotreater unit in sour water or get knocked out upstream of the fuel gas scrubber. If such knockout streams are sent to slop, water soluble chemicals can be drawn off from tanks and routed to wastewater treating or routed to crude units and end up in desalter water. Regardless of how the chemicals get into water they will end up in the wastewater treating plant.
DMDS and TBPS are both insoluble or only slightly soluble in water as are decomposition products dimethyl sulfide and butyl mercaptan. However, methyl mercaptan is soluble in water.
There is limited data on how much DMDS will not break down to H2S during sulfiding. It is clearly dependent on operating pressure, LHSV, type of catalyst and maybe other variables as well. As reactor temperatures are raised above 460-500°F for high temperature sulfiding, essentially all the DMDS decomposes to H2S.
There is some indication that sulfiding hydrocracking catalysts without hydrotreating catalysts, such as in a 2nd stage reactor, results in more of the DMDS sulfur getting into fuel gas as mercaptans. The reason for this is not clear but possibilities include:
-
Catalysts containing tungsten do not break down DMDS as well.
-
Zeolite catalysts do not breakdown DMDS as well.
-
Hyper-active sites that have not yet been titrated crack feeds even at initial sulfiding temperatures while temperatures are too low to hydrotreat these products, this implies the mercaptans come from sulfur in feed and not the sulfiding agent. On some occasions, mercaptan species that would not be created by breaking down sulfiding agents have been detected in fuel gas when sulfiding hydrocracking catalysts, this can be explained by assuming they come from hydrocracking feeds instead of decomposition of sulfiding agents.
-
There are several approaches that can be taken to minimize the impact of these mercaptans on fuel gas and sour water.
-
Reduce the rate that sulfiding chemicals are injected and sulfide one reactor at a time instead of multiple reactors at once. This will spread out the mercaptans in fuel gas over a longer time and reduce the peak impact. Unfortunately, this can extend the time to sulfide and get units up and running. Also, it won’t reduce the total amount of mercaptans sent to fuel gas, just the peak concentration. Plus, it will likely have minimal impact on mercaptans sent to wastewater treating.
-
Route the vents to fuel gas into the largest fuel gas system available. While this does not reduce the total SO2 in flue gas it will reduce the peak concentration.
-
Gas phase sulfide. This eliminates low pressure bleeds and may reduce the amount of mercaptans in contact with sour water. In a hydrocracker it also eliminates the risk of producing mercaptans from hydrocracking during sulfiding. However, it requires use of DMDS as TBPS cannot be used with gas phase sulfiding.
-
Use the highest purity make-up H2 available to delay the build-up of high concentrations of methane and heavier hydrocarbons in the recycle gas, this will minimize and possibly eliminate the need for a high-pressure gas bleed during the initial low temperature sulfiding. If a bleed is required, route the high-pressure bleed to another hydrotreater instead of to fuel gas. At normal operating temperatures, essentially all the mercaptans will be converted to H2S in hydrotreater reactors.
-
Use TBPS instead of DMDS. This has several advantages:
-
Since TBPS breaks down at lower temperatures than DMDS there may be less non-decomposed mercaptans at sulfiding temperatures.
-
Since byproducts are C4 mercaptans instead of C1 mercaptans they boil at higher temperatures and are less likely to be vented from either the high-pressure separator or from oil being circulated through low pressure equipment.
-
Using TBPS greatly reduces or eliminates the production of methyl mercaptan which is the only decomposition product with much water solubility, this should minimize mercaptans getting into the sour water treating plant.
-
Keep the Stripper and Charge Surge Drum as cold as possible to keep mercaptans in solution and recycle to the Reactor instead of venting. Also, keep stripping steam out of the Stripper. These steps also increase recycle of H2S to the reactor and reduce DMDS or TBPS injection requirements.
-
Route Stripper bottoms back to the Charge Surge Drum instead of to tankage to recycle mercaptans and H2S back to the Reactor. Mercaptans can be vented from tanks. If captured in a gas recovery system instead of going to atmosphere they will normally be routed to fuel gas.
-
Perform the low temperature sulfiding at a higher temperature than catalyst vendors recommend. Or if unwilling to do this, raise the first bed to around 460-500°F when the initial temperature wave reaches the second bed and use quench to keep the lower beds at recommended temperatures; this will increase mercaptan decomposition in the first bed after it has sulfur on it without the risk of reducing catalyst in the lower beds. Even if the sulfiding of the first bed is less effective this will often have minimal impact on the whole reactor especially if the top bed is used mostly for demetalization, olefin saturation and removal of the easiest sulfur species.
-
Caustic treats the vents to fuel gas to remove mercaptans.
-
Pre-sulfide the catalyst instead of sulfiding in-situ. This saves time during start-up and eliminates the need to deal with mercaptans. However, it is more expensive, requires obtaining the catalyst 1-2 months earlier to have time to pre-sulfide it and may mean inert entry is required in the reactor during catalyst loading.
The most significant steps that can be taken to reduce mercaptans getting into fuel gas will often be to either gas phase sulfide with DMDS and route high pressure bleeds to another hydrotreater or liquid phase sulfide with TBPS. Use of TBPS will likely be the most effective method of reducing mercaptans in sour water.
In the last several years Motiva has done a lot more presulfiding than in the past. Avoiding sulfur excursions in heater flue gas is one of the reasons we have been doing this.
SOREN MARKLUND (Chem32 LLC)
This may not directly address the issue at hand, but if wastewater mitigation is a serious issue, consider the use of pre-activated catalyst. In this case the hydroprocessing catalyst is delivered to the refinery already in its fully sulfided and activated state, meaning that activation inside the reactor will no longer be required. In this case, upon startup, there will be no water formation because of the sulfiding reaction. Since there will be no water formation, there will be no issues with contaminated wastewater, nor non-reacted mercaptan species. Starting up using pre-activated catalyst is comparably a very easy process, where the pre-activated catalyst is heated up under hydrogen/oil (or feed in gas phase if so required (i.e. Naphtha feed)). No more exotherms, no more requirements to hold at various plateaus, heating up rate only limited to the hydrogen embrittlement curve of said reactor. And, moreover, addressing this question, no foul-smelling water formation. CHEM32 located in Orange Texas can provide THIOCAT fully pre-activated hydroprocessing catalyst for your applications, and if cracked feed is present, THIOCAT CFT (cracked-feed tolerant) pre-activation can be provided to eliminate the 72h break in period under straight run feed, allowing starting up using cracked feed directly.
Year
2018
Process
Question 17: Where do your route your disulfide oil from caustic treatment? If disulfide oil, that may contain caustic or other contaminants, is routed to a hydrotreater, what are preferred methods for treating it to avoid fouling exchangers, furnaces and catalyst?
WENDY WILDENBERG (Flint Hills Resources)
We do not process directly from caustic treatment to a hydroprocessing unit either.
Disulfide oil is gravity settled out of the regenerated caustic in a settler vessel. A slip stream of cold Naphtha or #1 fuel oil is injected and used to help provide a larger volume of hydrocarbon to dissolve the disulfides and aid in separation (Sponge Oil).
The sponge oil / disulfide oil is sent to wet slop tankage where water from other wet slops can help to remove residual caustic. Wet slops use a floating suction line, like what Travis mentioned to transfer the oil to “dry” slop tank. Dry slop is routed to the coker fractionator where the slop oil is fractionated into coker naphtha, light coker gasoil, and heavy coker gasoil. Disulfide oil primary fractionates to the coker naphtha. No issues with fouling, spent catalyst poisoning are evident.
Year
2018
Process